您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-12-17 21:09
原料药质量是药品质量控制的关键和源头。其中,杂质的研究与控制事关药品的临床安全性,因而成为原料药质量控制的关键环节之一。药品临床使用中的不良反应除取决于药品本身的药理活性外,有时还与药品中的杂质密切相关,须严格控制。
从杂质来源分析,几乎不可能完全除去产品中的杂质,也没有必要。通过选择合适的分析方法,准确分辨与测定杂质含量,综合药学、毒理学及临床研究的结果,确定杂质的合理限度,通过对起始原辅料的源头控制、制备工艺的过程控制、包装材料、贮藏条件及有效期的确立等终端控制措施,将杂质控制在安全合理的范围内是杂质研究的最终目的。
可见,采用适当的分析方法实现各类杂质的检出与识别在杂质研究中居于重要的基础地位。
1 原料药杂质的分类与基本研究的思路
杂质是指在药品生产、运输、储存等过程中产生或引入影响药物纯度的物质,在人用药品注册技术要求国际协调会(International Conference on Harmonization,ICH)指南的Q3A(R2)中,将药品杂质分为无机杂质(inorganic impurities)、有机杂质(organic impurities) 和残留溶剂(residual solvents)3 类。
其中,无机杂质主要是指原料药制备过程中加入的催化剂、无机盐、配位体、试剂等;有机杂质主要是指中间体、副产物等工艺杂质以及药物本身降解、缔合或药物之间反应产生的有机物质;残留溶剂主要是生产中有机溶剂的残留。
杂质研究与控制的基本思路,是从杂质来源的分析入手,结合产品的实际生产工艺、结构特点等分析可能存在于产品中的中间体、副产物、降解物、反应物料等各种潜在杂质,通过杂质谱分析全面掌握产品的杂质概貌,根据各类潜在杂质的风险级别,有针对性地建立合适的分析方法,以确保各种潜在杂质的有效检出和确认。
跟踪杂质谱对安全性试验或临床试验结果产生的影响,并结合相关指导原则、文献信息等评估杂质的可接受水平,确立上市产品的杂质控制限度。
同时,通过杂质谱分析明确可能的杂质来源和去向,在制备工艺设置相应杂质的针对性控制措施,并通过产品包装和贮藏条件的研究,有效抑制药品的降解,实现从杂质产生的源头系统全面地把控药品杂质,从而保证药品的质量及安全性。
也就是说,杂质研究与控制的内容涉及杂质分析方法的建立与验证、杂质确认、杂质限度的确定及杂质控制诸多方面。
分析方法的建立与验证要结合杂质的不同特点对分析方法进行系统研究和验证,目的是使建立的分析方法适合于相应检测的要求;
杂质确认包括特定杂质与非特定杂质的确认、毒性(生物活性)杂质与一般杂质的确认和杂质谱分析等,目的是为杂质控制及限度确定提供依据;
杂质限度的确定是在综合杂质的特性、可接受水平、大生产能达到的水平基础上制订安全合理的限度;
杂质的控制是根据杂质研究结果,通过原辅料的源头控制(来源与质量)、制备工艺的过程控制(关键步骤与工艺参数)、稳定性控制(贮藏条件、有效期确定)等措施使杂质控制在安全合理范围。
2 原料药杂质研究与控制的基本现状及重点关注
在原料药的研究中,杂质的检出及控制至关重要。原料药的杂质是否能被全面准确的控制,直接关系到药品的临床安全性、质量可控性及产品稳定性。
所以,杂质研究是原料药质量保证的关键要素之一,同时也是国内外药品研发中重点关注、不断深入、快速发展的领域。随着人们对于药物研发规律认识的不断深入和分析技术的不断发展,杂质研究的理念逐渐系统化,监管措施也日臻成熟。
目前,国际上有关杂质控制的指导原则有:ICH 发布的Q3A(R2)、Q3B(R2)、Q3C(R5)和M7[8-9];欧洲药物管理局(European MedicinesAgency,EMA)发布的EMEA/CHPM/CVMP/QMO/450653/2006、CPMP/QWP/1529/04、CPMP/SWP/5199/02、EMEA/CHPM/QWP/251344/2006、EMA/CHMP/CVMP/QWP/199250/ 2009;
美国食品药品监督管理局(FDA)发布的NDAs、Genotoxic andCarcinogenic Impurities in Durg Substances andProducts(draft)、ANDAs ;澳大利亚药物管理局(TGA)发布的Impurities in Existing Drug Substances and Products (draft)等。
同时,中国药典2005 年版起即收载了杂质研究的相关指导原则,并于2005年发布了我国首个《化学药物杂质研究技术指导原则》和《残留溶剂研究技术指导原则》,这对我国药物研发中的杂质研究起到了一定的规范和指导作用。
随着对原料药杂质研究的不断深入,遗传毒性杂质、金属杂质、抗生素杂质等的检测及控制成为近几年原料药杂质研究与控制中的重点,并取得了重要进展。
2.1 基因毒性杂质
近年来,基因毒性杂质的研究与控制倍受关注,EMA、FDA 以及ICH 均针对此类杂质制订了相应的指导原则,为遗传毒性杂质的确认、研究和控制提供了指导性建议和技术要求。
尤其ICH 于2014 年7 月15 日正式发布的Draft Consensus Guideline Assessment and Control of DNA Reactive(Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk(M7),提供了可用于遗传毒性杂质鉴别、分类、定量分析和控制的可行性框架,系统回答了基因毒性杂质研究与控制中一些关键问题:
①如何根据结构活性分析评估杂质潜在的遗传毒性?
②如何确定杂质阈值(TTC)?遗传毒性杂质的可接受水平是多少?③为什么潜在基因毒性物质具有相似的分子结构?
④是否可以采用毒理学关注阈值(TTC)来规定遗传毒性杂质的水平?
⑤计算TTC 时,是否可以将多个遗传毒性杂质合并计算?
⑥哪些是可接受的特殊情况?日摄入量>TTC 的情况?
该指导原则根据风险级别将基因毒性杂质分为5 个类别:
①已知的致突变、致癌物。即致突变和致癌性数据为阳性,现有的公开数据库有ToxNet、NTP、InChem、BG‐Chemie、VITIC 和Leadscope 等。此类杂质应控制在化合物特定的限度以下。
②已知具有诱变性,但致癌效应未知,应控制在限度以下。
③含有警示结构,有潜在遗传毒性风险。但与原料药的结构无关,无致突变性数据。此类杂质应控制在限度以下,或进行细菌致突变性试验,如果细菌突变Ames 试验阳性=2类;细菌突变Ames 试验阴性=5 类。
④含有警示结构,与原料药和原料药结构相似物有关,但经检验并确认无致突变性。
⑤无警示结构,或含有警示结构,但有充分的数据证实无致突变性或致癌性。
此类杂质在很低的浓度下即可诱导基因突变并导致染色体的断裂和重排而具有潜在的致癌性,故研究者对这类杂质的检出聚焦在于痕量分析手段,重点选择专属性强、灵敏度高、重现性好及准确度高的分析方法。
目前报道较多的方法有气相色谱-质谱联用(GC-MS)、液相色谱-质谱联用(LC-MS)、毛细管电泳-质谱联用(CE-MS)、液相色谱-核磁共振-质谱联用(LC-NMR/MS)等,尤其是LC-MS 的应用最为广泛。
如Feng 等利用LC-MS 法同时检测甲磺酸伊马替尼中的甲磺酸甲酯、甲磺酸乙酯、甲磺酸丙酯3 个磺酸酯基因毒性杂质,其回收率分别为99.86%、101.7%、103.8%。刁荣蓉等建立LC-MS/MS 检测瑞替加滨的遗传毒性杂质,结果表明该方法可灵敏、快速、准确的测定其杂质含量。
2.2 金属杂质
近年提出“金属杂质”的概念正逐渐替代了此前定义不太明确的“重金属”概念,包括某些过渡金属和准金属,主要针对药物的合成和生产中试剂、催化剂、配位体等的残留以及与药物接触容器引入的金属杂质。
EMA、FDA 及ICH 相继颁布了金属杂质控制的相关指导原则,明确提出了金属杂质的分类和限度,ICH 还专门成立了金属杂质控制专家组,表明对此类杂质的控制已进入一个新的高度。
对于金属杂质,欧洲药典(EP7.0)、日本药典(JP16)、中国药典2010 年版均对重金属限度检查法有描述,但此方法类似于20 年前采用干燥失重方法检测有机溶剂残留量一样,存在误差大、专属性差、灵敏度低的缺陷,更无法有效控制高风险金属杂质残留量。
基于上述问题,2008 年美国药典(USP)发布了新的重金属控制专论(GeneralChapter),明确了多种金属元素的具体限度,对于重金属元素测定方法中的灵敏度、回收率、校准和漂移等均给出了相关规定,并对如何根据元素性质制备样品给出决策树。
但对于一些微量、少见的毒性金属元素仍没有给出控制限度。2014年5 月1 日正式执行的美国药典新通则<232>和<233>元素杂质控制新标准均予以补充。该标准明确要求测定各元素杂质的含量,即由之前的半定量转为定量控制,还规定了Cd、Pb、As、Hg、Ir、Os、Pd、Pt、Rh、Ru、Cr、Mo、Ni、V、Cu等15 种元素杂质的每日允许接触限值及原料药、辅料中金属杂质的限量值,并且第1 次明确了催化剂铂族元素Pt、Pd、Ru、Rh、Os、Ir 等的限度。
对于大容量注射液(注射剂量为10~100 mL),新标准规定,必须对各杂质在各组分中的限度分别求和,并建立限度值,制剂中各元素杂质限度值(μg·g1)用公式PDE≥[ΣM(CM×WM)]×DD求和。(M:制剂中各组分名称;CM:1 g 原料药或赋形剂组分含有的金属元素质量,单位:μg·g1;WM:1 个剂量单位中组分的质量;DD:最大日剂量)。
USP 将金属杂质分为2 类:一类为对人体会或很可能会造成毒性,以及造成严重的环境公害的金属,应严格控制不得出现;二类为毒性较一类低但应控制的金属杂质,具体分类和限度见表1。
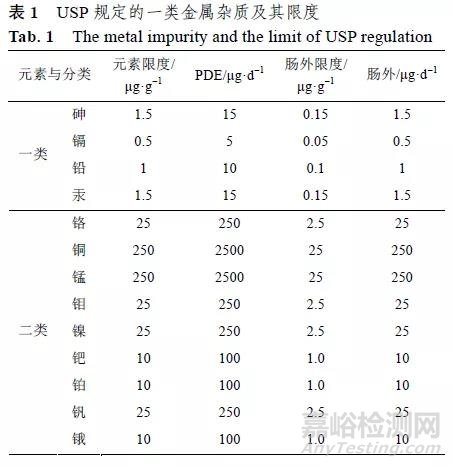
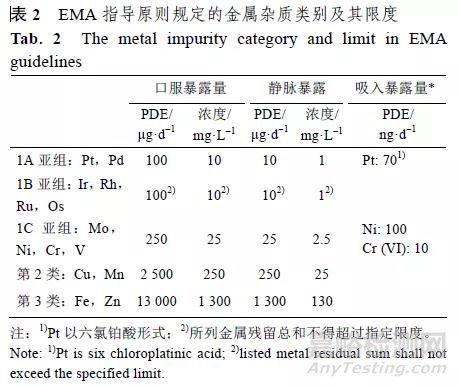
EMA 的指导原则没有说明USP 中的一类金属杂质,而是将USP 的2 类金属细分为3 个类别:第1 类金属为具有显著安全性担忧,具有已知或怀疑的致癌性其他显著毒性,包括1A 亚组:Pt、Pd,1B 亚组:Ir、Rh、Ru、Os,1C 亚组:Mo、Ni、Cr、V;第2 类金属为具有低的安全性担忧,具有潜在的较低毒性,如Cu、Mn 等;第3 类金属为安全性担忧最小,无明显毒性,如 Fe、Zn 等,具体见表2。
金属杂质的分类和限度控制对其分析方法提出了严格要求,传统的重金属检查法已远远不能满足此类杂质控制的要求,ICP-AES(电感耦合等离子体发射光谱)、ICP-MS(电感耦合等离子体质谱)以及GFAAS(石墨炉原子吸收分光光度法)越来越成为金属杂质研究与控制的常用方法。
与比色法相比较,可将比色法的半定量分析转为定量控制,克服了现行方法的局限性,大大提高了杂质检出的能力。
如Chen 等采用该方法检测格列齐特中的金属杂质,结果检测限<1 μg·g-1,甚至达到0.01 μg·g-1水平,而且回收率高达76.6%~123.0%。
如Zhao 等以Be 为内标元素校准基体效应及信号漂移,用ICP-MS 法直接测定那曲肝素钙中残留硼的量,硼的检出限为0.03 mg·kg-1,线性良好(r=0.999 8),RSD 为3.5%~5.1%,加标回收率在92.0%~96.2%。
2.3 抗生素杂质
抗生素一般通过生物合成(微生物发酵)、半合成方式得到。微生物发酵工艺具有明显不同于全化学合成工艺的特点,合成过程不是在反应中通过化学键的断裂与生成来实现,而是在微生物体内通过菌体的初级或次级代谢等生命过程完成。
这种生命过程是生物细胞按照其固有的遗传信息,在所处的培养条件下,进行复杂而细微的各种动态生化反应集合。
这些代谢过程难以与化学反应一样实施精细、准确的调控,只能在研究菌体的发育、生长和代谢等生命过程及各种生物、理化和工程环境因素对这些过程影响的基础上,对菌种、发酵条件和工艺过程的控制来把握产品质量。
与直接调控化学反应相比,这种“间接”的过程控制方式在把握药品质量方面具有相对较大的风险和难度,导致抗生素杂质谱较一般化学合成药更加复杂。
即使是半合成抗生素,起始物为微生物发酵产物,与一般的化学合成品相比,也具有纯度低、组份复杂、分子结构中活泼基团(如羟基、氨基、醛基等)较多、大多存在构型问题等特点。
因而导致多种副产物的产生,与纯化学合成产品相比,抗生素产品的杂质谱分析更复杂、更加难以预测和控制。
目前,除EMA 2010 年7 月发布了“Guideline on Setting Specifications for Related Impurities in Antibiotics”
外,ICH、FDA 及我国CFDA 均尚未发布抗生素药品杂质研究的专门指导原则。
EMA 指导原则提供了关于发酵或半合成的抗生素中有关杂质报告限度、鉴定限度和确认限度的方法,详见表3。
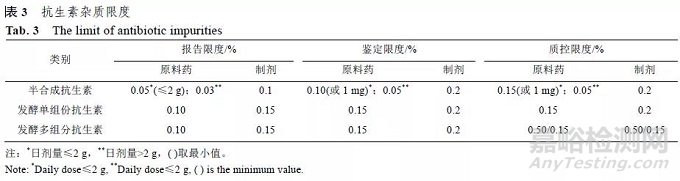
按照该指导原则,与母体化合物结构密切相关的有关物控制限度为0.50%,其他有关物质的控制限度为0.15%。
如果申请人声明有关物质(未包含在活性物质中的化合物)与母体化合物密切相关,必须采用HPLC/MS 分析法或HPLC/二极管阵列检测器或使用分析标识物予以证实;
杂质分析时只要可能,应使用外标法来计算重量比,以便评估和排除任何可能的质量不守恒;
如果使用面积归一法,相关成分和有关物质应在检测器中产生相似反应,否则应使用响应因子进行校正。该指导原则所述杂质不包括发酵工艺中产生的残渣,如来自培养基、产物母体和微生物发生器的残渣等。
Liu 等通过对比中国药典、美国药典和欧洲药典中对头孢丙烯原料杂质的控制标准与方法。发现美国药典未对头孢丙烯有关物质检查做出要求,但在含量测定中规定了头孢丙烯(Z)和(E)异构体的控制标准;
中国药典除规定头孢丙烯(Z)和(E)异构体外,还规定了头孢羟氨苄杂质限量和其他杂质总限量;欧洲药典除规定头孢丙烯(Z)和(E)限量外,还规定了另外14 种杂质的相对保留时间和限量标准,并给出了它们的结构式。
可见,欧洲药典对头孢丙烯质量控制标准最为严格和具体。
3 杂质分析方法的新进展
分析方法是获取杂质信息的手段,直接关系到对药物杂质信息把握的准确性和全面性。因此,杂质研究的首要问题是选择合适的分析方法。
对于原料中杂质的检测,应根据药物及杂质的理化性质、化学结构、控制要求选择适当的方法检测。杂质的微量性和多样性决定了具有分离分析特点的色谱法成为杂质研究的常用方法, 主要有HPLC、GC、GPC 和CE 等。
但是,单一方法均存在一定的局限性,所以对杂质进行分析时,应运用不同原理的分析方法相互补充与验证。
目前,国际前沿的杂质研究与分析不断吸纳当今分析科学新成就和药物研发规律新认识,色谱仪器越来越专业化(凝胶色谱、离子色谱、逆流分溶色谱等相继用于有关物质检测),色谱与光谱联用技术越来越成熟(HPLC-UV,HPLC-MS,HPLC-NMR;GC-MS,GC-IR 等联用仪器正在普及应用中),各类数据库越来越丰富,联机智能化解析系统越来越普及,为杂质研究提供了更为完善的利器。
新型色谱填料和不同类型检测器不断出现在药品的杂质控制中,USP 自第26 版开始,每版都有新类型的色谱柱增加,USP 第31 版即已增加至63 种固定相填料,使得采用多样化的色谱条件和检测手段(HPLC-UV/ECD/ELSD/RID)有效控制不同类型杂质成为可能。
杂质检测逐步呈现出全面检出(梯度洗脱,多检测手段互补、明确灵敏度要求)、有效分离(规定最难分离物质对峰间的分离度)、目标明确(特定杂质、非特定杂质)、准确定量(外标、校正因子、归一法相结合)的趋势。如Shen 等利用HPLC-DAD/MS对辛伐他汀中潜在的7 个已知杂质A、B、C、D、E、F、G 进行了归属分析,弥补了高效液相法保留时间定位的不足。
LC-MS 联用技术将LC 的高分离能力和MS的高灵敏度、高专属性融于一体,主要分为高分辨率质谱(HRMS)、多级质谱(MSn)、氢/氘转换质谱(HDE-MS)3 类。
在原料药的杂质谱分析中,可鉴别已知杂质和推导未知杂质的结构。
如Li 等[30]采用超高效液相- 三重四级杆质谱联用技术(UPLC-MS/MS)研究辛伐他汀的杂质谱,推出了含量≥0.05%的12 个杂质的结构。
杨丽珍等运用高效液相色谱-四极杆飞行时间质谱法分析克拉屈滨的主要杂质及来源。
HaRigaya 等利用HPLC-ICP-MS 检测安替比林中残留苯肼杂质。LCMS联用技术还被广泛用于检测原料药储藏过程中产生的降解产物,为药物的储藏、运输及整个过程中的存放条件提供理论依据。
总之,LC-MS联用技术结合杂质谱的“指纹”特征,可以快速评估合成工艺,并对杂质结构进行归属,对原料药杂质的控制具有指导作用。
GC-MS 联用技术具有高选择性、高灵敏度及含有丰富的结构信息等优点,特别适合易挥发的多组分混合物中未知组分的定性、定量分析,对有机挥发性杂质和残留溶剂的测定是不可或缺的工具。
如Zhang 等利用GC-MS 对磺酸酯类物质的痕量分析,有效的控制了杂质的限度。Razboršek等考察了反式迷迭香酸在光照、高温、不同溶剂和湿度条件下的稳定性,并利用GC-MS 分析反式迷迭香酸的降解产物,通过MS 碎片离子来推导降解产物的化学结构,结果显示其唯一降解产物为顺式迷迭香酸。
LC-NMR 联用技术具有专属性强、准确、灵敏等优点,常被运用于未知杂质的研究。
如Pan等使用LC-NMR 联用技术鉴别TCH346 中的降解杂质。Murakami 和Provera 等将LC-NMR、LC-MS 运用于NK1 拮抗剂GW597599 原料药中杂质结构的鉴定。
Novak 等利用停留模式的LC-NMR 联用技术快速鉴定新型抗真菌药Icofungipen 的未知杂质,并合成了该杂质。
CE-MS 联用技术适合用于分析在溶剂中易解离的强极性化合物。有一些研究者采用具有较高的检测灵敏度且操作简单的在线推扫富集技术补充其不足。如Zhao 和Du 等利用胶束毛细管电泳在线推扫富集技术测定了乳酸环丙沙星原料中的痕量杂质,并解决了胶束毛细管电泳在测定过程中重现性差的问题。
4 结语
药品中的杂质是否能被全面准确地控制,直接关系到药品的质量可控与安全性,原料药的杂质研究与控制是保证药品安全性和质量可控性的前提和关键环节,是药物一致性评价的重要指标。
根据掌握的杂质谱概况,依据各类潜在杂质的风险级别、产生的可能性高低制定进一步的研究控制策略。
杂质的控制一般应包括每个明确的已知杂质、每个明确的未知特定杂质、任何非特定杂质(不超过鉴定限度)以及总杂质,关注是否进行了高毒性杂质与一般毒性杂质、毒性杂质与一般杂质、新增杂质与超量杂质、特定杂质与非特定杂质、单个杂质与总杂质的研究与控制。
当试验表明现有技术的确无法鉴定某个杂质时,至少要提供此杂质结构的充分证据来表明它可归属为母体化合物或某侧链等有关物质,将其作为明确的未知杂质使用适当的分析标识手段进行识别和控制。
遗传毒性的控制是目前杂质研究中的一个新课题,也是药品研发中风险控制的关键把手,研发中需要根据有机化学反应机制分析原料药合成、纯化、制剂生产和贮存过程中很有可能产生的实际的和潜在的遗传毒性杂质,依据相关毒性物质数据库甄别、评估可能具有基因毒性的杂质或具有基因毒性结构单元的杂质,参照国内外相关技术文件的研究思路与策略,对此类杂质进行针对性的确认、检出和控制,充分保证产品的安全性。
作者:郝杰

来源:《中国现代应用药学》


