您当前的位置:检测资讯 > 热点事件
嘉峪检测网 2022-05-04 22:06
昨日,和黄医药发布公告称,已收到美国FDA有关索凡替尼用于治疗晚期神经内分泌瘤的完整回复函。FDA认为当前基于两项成功的中国III期研究以及一项美国桥接研究的数据包尚不足以支持药品现时于美国获批。该完整回复函中表明,需要纳入更多代表美国患者人群的国际多中心临床试验。

未获批,需补充国际多中心三期临床试验,似曾相似的审判彷佛就在昨天。
今年2月11日,信达生物发布公告称,美国FDA召开肿瘤药物咨询委员会(ODAC),对信迪利单抗注射液的新药上市申请(BLA)审评问题进行讨论并投票。最终,委员会14:1投票建议需要在获批前补充额外临床试验,证明信迪利单抗在美国人群和美国医疗实践中的适用性。
被同一块石头绊倒两次,我们无法得知这其中是否有什么隐情,但从之前的公告中可看出一丝端倪。
2020年5月,和黄医药称,在新药上市申请前会议上已与FDA达成一致,认为索凡替尼用于治疗胰腺和非胰腺神经内分泌瘤患者的两项取得积极结果的中国III期研究,连同索凡替尼美国桥接研究的现有数据,可构成支持在美国提交新药上市申请的依据。
FDA的这个“甜枣”,信达也不能说没收到过。
“FDA审批改革风向标”,美国FDA肿瘤学卓越中心(OCE)主任理查德·帕兹杜尔(RichardPazdur),曾在2019年4月美国癌症研究协会(AACR)年会上高调表态:美国欢迎中国的PD-1,哪怕仅有中国本土的临床数据,只要质量好的话,FDA肯定为其敞开大门。
失望也好,遗憾也罢,终是到了梦醒的时候。我们无从得知FDA前后不一的表态是制药公司单方面一厢情愿,还是FDA审评标准不断与时俱进;但可以确定的是,仅依靠中国中心的临床试验就想敲开美国FDA的大门,终究是一场不切实际的大冒险。想在别人的地盘上赚钱,还得先按照别人的规则来,毕竟主动权在FDA手里。
突破性疗法/快速通道
从2017-2021年每年FDA获批的药物可见,快速通道和突破性疗法都占据到了不小的比例,体现了药物早期差异化数据的重要性。
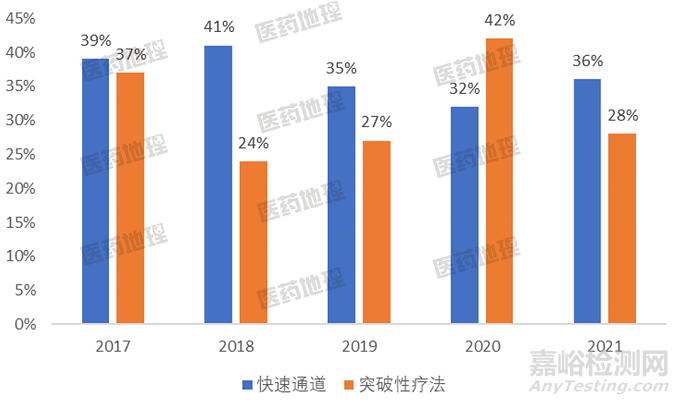
图1: 2017-2021FDA批准药物中突破性疗法和快速通道占比情况
快速通道旨在帮助开发药物并加快对药物的审查,这些药物在治疗严重或威胁生命的疾病方面显示出希望,并能满足尚未满足的临床需求。
证明未满足临床需求所需的信息类型随药物开发阶段的不同而不同:在开发初期,非临床数据,机理论据或药理学数据就足够了;在以后的开发中,应利用临床数据。如果有现有疗法,那么快速合格的药物必须显示出优于现有疗法的优势,例如:
(1)显示出卓越的功效
(2)避免严重的副作用或者致停药的副作用
(3)针对正在出现或预期出现的公共卫生需求等。
突破性疗法与快速通道的差别在于,“突破性疗法”旨在加速开发及审查治疗严重的或威胁生命的疾病的新药。其初步的临床证据表明有一项临床有意义的终点指标较现有药物有实质性改善(Substantialimprovement)。对于实质性改善的界定,FDA表示需要同时考察临床治疗维度(如药物效果持续的时间)以及临床疗效维度(比如疾病缓解率ORR)等,即在初步临床数据中已经能够看到清晰明确的优势。
对于美国FDA突破性疗法而言,2012-2021年十年间共有470项得到了相关资格认定,其中已经有240项成功获批上市,考虑到仍有较多获得认定的尚处于临床和上市申请情况,获得突破性疗法资格的药物其上市于成功率将高于50%。根据Naturereview drugdiscovery的报道,2010-2017年获选药物从进入临床一期到获批的概率约为6%-7%,从进入临床二期到获批的概率约为11%-15%,突破性疗法授予的时间段为不晚于临床二期结束,由此可见获得初步数据认可的突破性疗法有望体现出更高程度的成功率。
FIC(Firstin Class)药物
FIC(FirstinClass)药物不一定能成为重磅药物,但成为重磅药物的大部分是FIC药物。根据FDA定义,被授予FIC的药物是指使用全新的、独特的作用机制来治疗某种疾病的药物。FIC药物又可进一步分为四类:
(1)靶点(组合)新,适应症新,该类药物如果能够展现不错的疗效,较为容易获得突破性治疗等认定,注册临床往往也可采用单臂的方式;
(2)靶点(组合)老,适应症全新,注册临床方面参考(1),但需要充分的概念验证,特别是在相关靶点的其他药物并没有对该适应症作用的情况下,需要谨慎设计和判断;
(3)靶点(组合)新,适应症老,该类药物在注册临床方面往往需要和该适应症的SOC进行头对头非劣效试验,当然,如果在早期临床中相关药物已经能够展现出与现有SOC比明显的优效,依旧有可能以突破性疗法的形式快速获批;
(4)靶点(组合)老,适应症在该靶点药物方面为首创,需要同时考虑(2)和(3)的情况,面临的市场空间可能也相对有限。
临床明确获益的产品
将明确临床获益放在最后并不是因为其不重要,而是其重要性已得到了普遍认可。
西达基奥仑赛(cilta-cel)是近期创新药出海的硕果仅存。3月1日,金斯瑞发布公告,由强生和传奇生物合作开发的BCMACAR-T产品西达基奥仑赛,已获得美国FDA批准上市,用于治疗复发/难治性多发性骨髓瘤成人患者(r/rMM)。
2017年,在美国临床肿瘤学会(ASCO)的年会上,传奇公布了当时的代号还是LCAR-B38M的西达基奥仑赛,治疗复发难治性多发性骨髓瘤病人,100%总体缓解率。虽然只是早期临床数据,但足以让世界为之震惊。
多发性骨髓瘤发病率为2~4/10万,男女比例为1.6:1,常见于中老年男性患者中。尽管过去十几年,创新疗法的不断出现革新了多发性骨髓瘤的治疗方式,然而很多患者在获得缓解之后疾病仍然会复发,并且对已有疗法产生耐药性,最终进展为复发难治性多发性骨髓瘤,危害患者生命。作为一次性输注疗法,西达基奥仑赛能够为该患者群体提供持久深度的缓解,具有提高长期生存率的潜力,为持续接受治疗的患者提供转机和希望。
和黄药业本次出海失利,在某种程度上其实是为创新药出海扫清了阴霾。脚踏实地,勇于创新,解决未满足的临床需求,没有捷径就是最好的捷径。

来源:医药地理


