您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-08-07 20:00
一些制药公司通过各种创新技术来提高开发新药的速率的过程已变得日趋艰难.即使公司投入巨大的资金用于技术更新,但是其销售额仍远远跟不上新药研究和开发所需费用的迅速增长。伴随着上述问题的出现以及全球医药价格上的竞争、基因时代的挑战等。医药企业开始重视Me-too策略在新药研究开发中的应用。实验事实表明,通过对Me-too策略来发现新药确实可以极大地提高开发新药的速率,越来越多的公司开始应用Me-too策略对现有的药物分子进行修饰和改造,目前已有许多成功实例。
1H2受体拮抗剂的Me-too研究
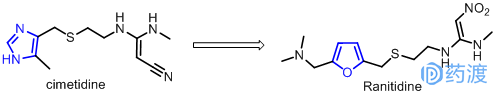
当年为寻找抑制胃酸分泌、治疗消化道溃疡药物,美国史克公司根据H2受体拮抗剂理论,花费不少心血。耗时十多年,成功开发了西咪替丁(Cimetidine),其化学结构由咪唑五元环,含硫醚的四原子链和末端取代胍3部分构成,成为第一个高活性的H2受体拮抗剂药物,于1976年在英国率先上市。它一问世很快成为治疗溃疡病的首选药,到1979年在l00多个国家获得上市许可。西咪替丁的问世,开辟了寻找治疗溃疡病的新领域。葛兰素公司的科学家们得知关于H2受体拮抗剂有研制成功新药的可能性后,曾一度假定咪唑环是该类药物与H2受体识别的必要结构,早期的改造工作集中在侧链的变化上,但未得到比西咪替丁更优秀的药物。后来用呋喃环代替了组胺的咪唑环,于1981年推出雷尼替丁(Ranitidine)。1983年雷尼替丁成了第二个上市的H2受体拮抗剂,其疗效是西咪替丁的5~8倍,对胃和十二指肠疗效高,且有速效和长效的特点,其副作用较西咪替丁小,无抗雄性激素的副作用,且与其它药物的相互作用也较小。由此其成为世界目前最畅销的药品,年销售量达30亿美元,连续几年排在世界畅销药的首位。
2血管紧张素转化酶抑制剂的Me-too研究
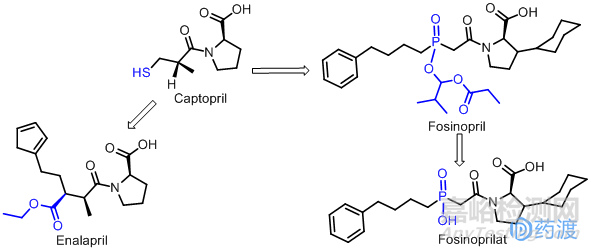
血管紧张素转化酶(ACE)抑制剂的开发,大量的早期工作是在大学完成的。当时并没有引起人们的商业追求,直到20世纪70年代后期,才成功开发出了卡托普利(Captopril),于1981年在美国首次上市。血管紧张素转化酶有一个锌离子,而巯基(-SH)是与锌离子结合的部位,但卡托普利也因带有巯基而略带有大蒜的气味。Merck随后在1984年推出了依那普利(Enalapril),该品种由于去掉了分子中的巯基,含有酯基成为前药,可以改善吸收,能进入中枢,在体内水解,游离出的羧基可选择性地与活性中心亲和,增强了与酶结合的强度,治疗作用比卡托普利强10倍,且毒性小,作用持久。依那普利在1995年全球销售额达25亿美元。20世纪90年代,B-M Sqllibb公司又推出了福辛普利(Fosinopril),是一种含磷的新型血管紧张素转换酶抑制剂,为前体药,对ACE直接抑制作用较弱,但口服后缓慢且不完全吸收。并迅速转变为活性更强的二酸代谢产物福辛普利拉(Fosinoprilat),作用持久。福辛普利拉通过其次磷酸基团和ACE活性部位中锌离子的结合,抑制ACE活性,福辛普利对肾功能无明显影响,成为第三代ACE抑制剂的代表。
3离子泵抑制剂的Me-too研究
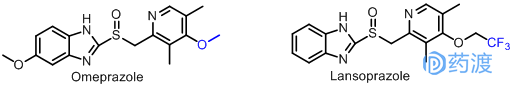
奥美拉唑(Omeprazole)是Astm公司精心研制的第一个质子泵抑制剂,于1988年上市,该药对胃酸有强而持久的仰制作用。而兰索拉唑(Lansoprazole)是奥美拉唑的衍生物,它有奥美拉唑一样抗酸分泌的作用,但是它的稳定性和口服利用度较奥美拉唑都有显著提高。从结构上看,兰索拉唑与奥美拉唑除了一个氟取代的烷基外,几乎完全相同。
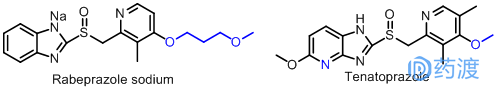
近年来,国内外的许多厂家也根据奥美拉唑和兰索拉唑的结构以及“Me-too药”的研究方法生产出了一大批新药。如1998年在日本上市波利特(雷贝拉唑钠,Rabeprazole sodium),于1999年引入欧洲,同年经美国FDA批准在美国上市,带来了全新的治疗选择短时间内就得到了国外广大医师的认可,为众多消化性溃疡和胃食管反流病患者,从结构比较上可以看出是奥美拉唑的结构类似物,其结构与兰索拉唑极其相似,只是侧链稍作改变,将原来的三氟乙氧基换做了甲氧丙氧基,其他部分基本一样,也属于“Me-too药”的一种。2004年在日本上市的泰妥拉唑(Tenatoprazole)是由日本东京田边、日本三菱公司和日本北陆制药公司联合研制开发的一种新型胃H+/K+-ATP酶抑制剂(即质子泵抑制剂)。该药显著抑制胃酸的分泌,同时对幽门螺旋杆菌也有抑制作用。疗效比奥美拉唑强7倍,而且稳定性也较奥美拉唑有显著提高。就其结构来看,就是将奥美拉唑结构中的苯环用吡啶环替换而生产出另一新药。
4二氢吡啶类钙拮抗剂的Me-too研究
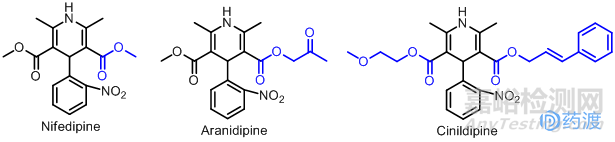
二氢吡啶类钙拮抗剂是一类特异性高、作用很强的药物,具有很强的扩血管作用,适用于冠脉痉挛、高血压、心肌梗死等。最早使用的是硝苯地平(Nifedipine),人们根据构效关系的结构修饰合成出一系列的“地平(dipine)”类化合物:阿雷地平(Aranidipine)是由日本Taiho公司于1996年上市一种二氢吡啶类新药。它是在硝苯地平的基础上加以修饰将二氢吡啶环的3位上酯基上的甲基氢用乙酰基替换而得。该药能够立体选择性地抑制钙通道,且与受体结合和解离的速度较慢,降压效果较硝苯地平更为持久。西尼地平(Cinildipine)是由日本富士株式会社1996年上市的长效二氢吡啶类钙拮抗剂,它是对硝苯地平3,5位的酯基进行替换所得,其结构中酯基的反式双键对钙通道有阻碍作用,也是一种长效的降压药。后两种药物都是根据硝苯地平的结构进行一定改变而得的新药,都是Me-too药的经典代表。
5羟甲戊二酰辅酶A还原酶抑制剂的Me-too研究
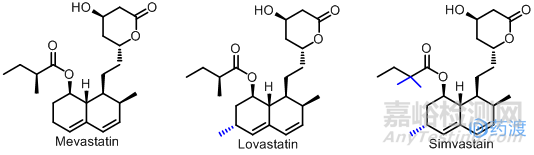
在20世纪70年代初,日本北里医药研究所的Endo等几位微生物学家一个偶然的机会发现了一种未知物能抑制羟甲戊二酰辅酶A(HMG-CoA)还原酶的活性,从而明显地降低血浆中的胆固醇。日本科学家将这种新发现的物质命名为康帕丁(Compactin),即美伐他汀(Mevastatin)。美伐他汀的发现开启了寻找和发展HMG-CoA还原酶抑制剂类调节血脂的新纪元。但是由于当时的工业化技术未完全过关,加上日本医药企业也没有在美伐他汀及其他衍生物产品上投人大的资金继续深入开发下去,最终未能形成生产能力,并失去了他汀类调节血脂药的开发优势。
然而日本学者有关他汀类血脂药物的研究成果引起了西方国家医药界同行的强烈兴趣。美、德、英、法、瑞士等老牌的制药工业强国在他汀类药物研究开发方面投入大量的人力和财力,并取得了丰硕的成果。在不到20年的时间里西方各国共计开发了包括美伐他汀在内的10多个他汀类调节血脂药物。由默克公司开发的洛伐他汀(Lovastatin)于1987年首次在美国上市成为第一个上市的他汀药物,是一种无活性的前药,需在体内将酯环水解成开链的羟基酸衍生物才有抑酶活性,该药能竞争性抑制HMG-CoA还原酶,降低胆固醇的合成及载脂蛋白的浓度;增加低密度脂蛋白受体的活性;还少量降低血浆中的甘油三酯及极低密度脂蛋白中胆固醇的浓度,升高血清高密度中的胆固醇水平;与降酯药合用有良好的协同作用.继洛伐他汀之后又成功开发了辛伐他汀(Simvastain),它是洛伐他汀的甲基化衍生物,其活性比洛伐他汀强1倍,Merck公司1981年在欧洲、1984年在美国申请专利。
随后又产生了一系列的Me-too药物,如日本三共公司开发的普伐他汀、瑞士山道士公司开发氟伐他汀、德国拜耳公司开发西立伐他汀以及美国华纳一兰伯特公司(现并入辉瑞公司)开发阿托伐他汀等。
6生物烷化剂中氮芥类药物的Me-too研究
氮芥类药物的发现源于芥子气。芥子气是一种烷化剂毒剂,第一次世界大战期间被作为毒气使用,后来发现芥子气对淋巴癌有治疗作用。由于其毒性太大,不可能药用,就在此基础上发展出氮芥类抗肿瘤药,于是以N原子代替S原子成为氮芥类药物的改造重点。

从氮芥类结构上来说,R是载体部分,可以改善该类药物在体内吸收、分布、等药代动力学性质,其他部分属于烷基化部分时抗肿瘤活性的功能基。当载体部分是脂肪烃基时成为脂肪氮芥,是芳香环取代时则为芳香氮芥。
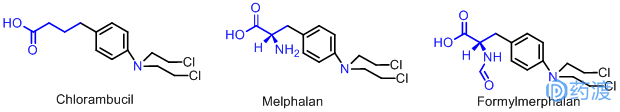
芳香氮芥类构效关系研究表明当羧基和苯环之间碳原子之间原子数为3时效果最好,即苯丁酸氮芥(Chlorambucil,瘤可宁),用于治疗慢性淋巴细胞白血病,对淋巴肉瘤、霍奇金病,卵巢癌也有较好的疗效,临床上用其钠盐,水溶性好,以被胃肠道吸收,在体内迅速转化为游离的苯丁酸氮芥。
另一个成功的例子是在芳酸的侧链上进行取代,引入天然存在的氨基酸,以期增加药物在肿瘤部位的浓度和亲和性,提高药物的疗效。例如以苯丙氨酸为载体的美法仑(Melphalan,溶肉瘤素),对卵巢癌、乳腺癌、淋巴肉瘤和多发性骨髓等恶性肿瘤有较好的疗效。
中国学者在美法仑基础上将NH2进行甲酰化得到氮甲(Formylmerphalan),与溶肉瘤素比较,治疗指数高,毒性较低。
在我国专利制度日益完善、新药研发资金投入相对不足、研究水平相对落后的现实情况下。采用Me-too策略发现新药,相对于新药创新来说,降低了技术难度、风险和研发成本,这样既可以避免专利侵权,又可以加快我国化学合成药物的研究。为新药研究开发工作提供必要的技术积累和资金积累,促进我国医药行业的发展。

来源:药渡


