摘 要 Abstract
本文梳理了医疗器械创新产品注册程序实施以来,在我国上市的24 个进口创新医疗器械的时间,发现与美国上市的时间差平均为4.4 年,这与国家提倡的尽快引入先进产品、鼓励全球同步上市的目标尚存在差距。为研究进口创新医疗器械在我国同步上市的影响因素,本文整理了我国创新医疗器械资质申请、临床试验备案和审批以及注册申请的流程与要求,并与美国突破性医疗器械的上市路径进行对比。结果表明,专利申请、型式检验以及临床试验审批是最易导致上市时间滞后的环节,进而影响我国加入全球多中心临床试验和同步研发。本文提出延长创新医疗器械资质时长、规范医疗器械临床阶段变更路径、统一临床试验审批中对境内外企业的要求、加快国际标准本地转化、完善专利制度、允许创新产品自主灵活定价、鼓励境内企业开展海外合作等几点建议,以期为进口创新医疗器械在我国同步上市政策研究提供思路与借鉴。
This paper reviews the time of 24 imported innovative medical devices approved in China since the implementation of the innovative medical devices registration procedure, and finds the average time lag is 4.4 years compared to their market launch in the United States, indicating a gap from the national advocacy of promptly introducing advanced products and encouraging global simultaneous market launches. To investigate the factors affecting the simultaneous market entry of imported innovative medical devices in China, this article outlines the processes and requirements for innovative medical device designation application, clinical trial filing and approval, and market application for innovative medical devices. A comparison is made with the marketing pathway for breakthrough medical devices in the United States. The comparison reveals that patent application, type testing, and clinical trial approval are the key stages that most likely contribute to the time lag in market approval, impeding China's participation in global multicenter clinical trials and collaborative research. Therefore, this article proposes relevant policy considerations and recommendations, including extending the designation validity duration of innovative medical devices, standardizing the pathways for device changes during the clinical stage, unifying the requirements for domestic and foreign applicants in clinical trial approvals, accelerating the local translation of international standards, improving the patent system, allowing innovative products set prices independently, and encouraging domestic enterprises to engage in overseas collaborations in multiple ways.
关键词 Key words
创新医疗器械;同步上市;临床试验审批;型式检验;突破性医疗器械
innovative medical device; simultaneous marketing; clinical trial approval; type testing;breakthrough device
深化医药卫生体制改革是建设健康中国行动的重点内容,其中加速药品和医疗器械的审批上市是建设健康中国、深化医疗卫生领域供给侧结构性改革的重要举措。促进以临床需求为导向的创新药品和医疗器械在我国上市,有利于扩充现有诊疗手段,丰富治疗方案。《中华人民共和国国民经济和社会发展第十四个五年规划和2035 年远景目标纲要》中,明确提出了“完善创新药物、疫苗、医疗器械等快速审评审批机制,加快临床急需和罕见病治疗药品、医疗器械审评审批”[1],强调了对技术创新产品和临床急需产品的特别关注。在《“十四五”医药工业发展规划》中,进一步对医药产业的创新能力及国际化地位进行了展望,提出要吸引全球医药创新要素向国内集聚,包括吸引全球创新药品和医疗器械率先在我国注册,整体缩短创新产品国内外上市时间差,促进临床急需的境外已上市新药和医疗器械尽快在境内注册[2]。2021 年,国家药监局等8 部门联合印发的《“十四五”国家药品安全及促进高质量发展规划》聚焦在产品审评审批的目标上,提出“加强创新产品审评能力,能够同步审评审批全球创新药物和医疗器械,支持境外新药和医疗器械在境内同步上市”[3]。由此可知,如何使我国公众尽早用上国际先进医疗产品已成为我国药品监管部门关注的重点之一。
让国际先进医药产品在我国同步上市对于医药行业全链条的发展都具有促进意义:①满足临床需求,使患者不必万里求医,可同步分享全球医药创新成果;②吸引全球医疗资源向我国汇集,提高我国患者和市场的关注度;③弥补诊疗空白,学习国际最先进的治疗方案,提高我国医药从业者在国际上的话语权和参与度;④提高监管机构的创新产品评价能力,逐渐与国际监管、标准接轨;⑤提高市场活力,以进口竞争带动我国医药产品研发水平进步,促进全球技术交流,有利于国产产品走出国门。
目前,我国药品领域已有产品基本实现了全球同步上市,例如勃林格殷格翰公司研发的佩索利单抗注射液和艾伯维公司研发的乌帕替尼缓释片,我国与美国同步研发,并参与国际多中心临床试验,实现了同步申报以及同步上市。然而,进口医疗器械领域目前罕有同步上市情况。因此,本文探讨了进口医疗器械全球同步上市现状,以期探究影响我国进口医疗器械同步上市的因素,并提出相应政策建议。
1、 同步上市的路径与现状
关于同步上市的定义在我国现有法规中尚未有明确的表述,一般可以理解为产品在我国境内和境外的上市时间差尽可能小。考虑到各国和地区审评时限不同,同步申报也可以划入同步上市的范畴。
若要缩短进口产品上市的“时间差”甚至是争取同步上市,就必须利用好特殊注册程序。《医疗器械注册与备案管理办法》中,规定了3 种特殊审批程序可以加速医疗器械的上市审批。3 种路径的适用场景各有侧重:①创新产品注册程序,侧重对有核心技术发明专利、有显著临床优势且国际领先的首创产品的扶持。②优先注册程序,侧重临床急需、国家研发重点及其他情形下的优先审查。③应急注册程序,侧重对突发公共卫生事件的应对[4]。
值得注意的是,《医疗器械监督管理条例》第十六条规定“未在境外上市的创新医疗器械,可以不提交注册申请人所在国(地区)主管部门准许该医疗器械上市销售的证明文件”[5]。也就是说,其他途径都需要该产品在原产国(地区)获批后,将原产国(地区)上市批件作为在境内申报的递交文件之一,只有创新医疗器械最可能实现境内外同步上市。因此,创新医疗器械的申报路径为本研究的重点。
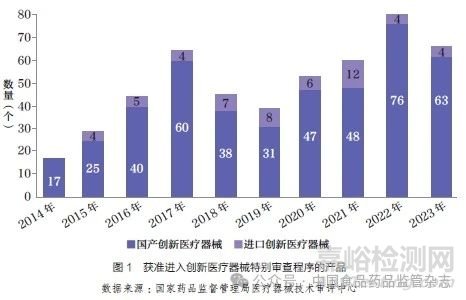
截至2023 年, 我国共有499 个产品获准进入创新医疗器械特别审查程序,其中54 个为进口产品,约占10.8%,如图1所示。
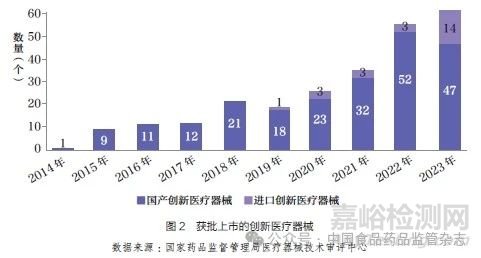
按照“标准不降低,程序不减少”的原则,国家药品监督管理局医疗器械技术审评中心(以下简称器审中心)对创新医疗器械产品给予了早期介入、专人负责、加强沟通等帮助,较其他第三类医疗器械首次注册的平均时间缩短近3 个月[6]。截至2023 年,获得创新医疗器械资质的产品中,已有250 个产品获批,其中进口医疗器械24 个,约占9.6%,如图2 所示。
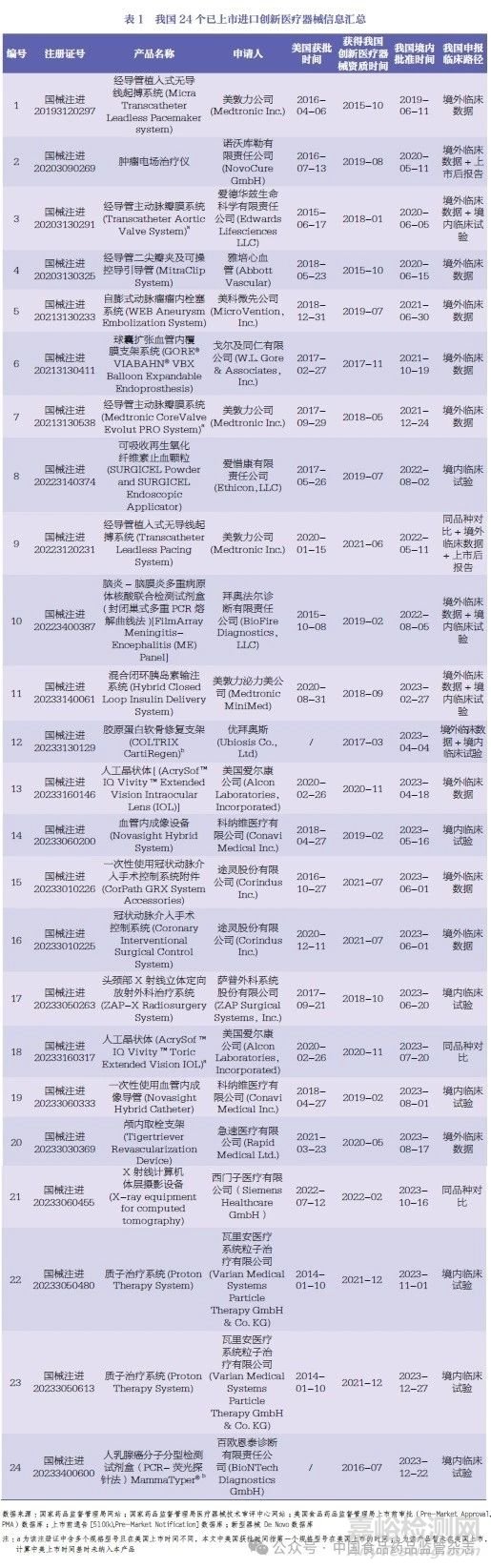
在已上市的24 个进口创新医疗器械中,与美国上市时间差最短的是西门子医疗有限公司的X射线计算机体层摄影设备,在美国上市461 天后在我国上市。这24 个进口产品中有22 个已在美国上市,我国与美国上市的平均时间差是4.4 年,与《“十四五”国家药品安全及促进高质量发展规划》中提出的境内外同步上市的目标尚存在差距。
本文汇总了我国已上市的24 个进口创新医疗器械产品的基本信息以及在美国的获批时间,见表1。需要说明的是,这24 个产品不一定在美国首先上市,可能选择了在欧盟、研发地或生产地申请上市。本文只列举了这24 个产品在美国的上市时间。一是因为美国作为全球医疗器械的主要市场,产品获得FDA的批准具有参考意义。二是笔者希望通过与美国突破性医疗器械程序(Breakthrough Medical Device Program) 进行对比,分析其优势,以获得提升我国相关规定的启示。
2、 上市流程的挑战
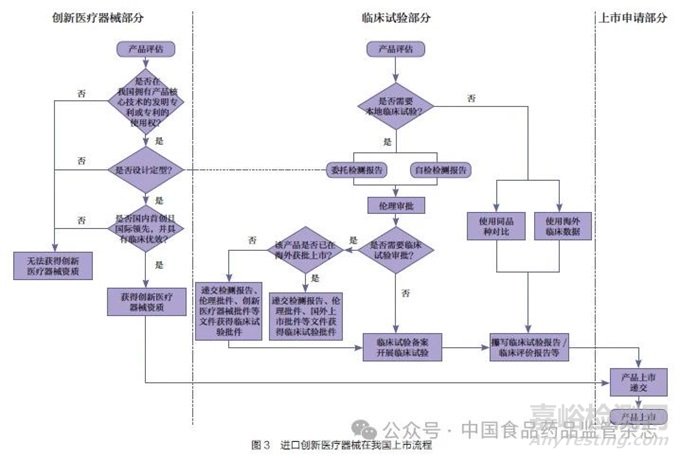
本文梳理了进口创新医疗器械在我国上市的全部流程,包括资质申请、临床路径以及上市申报递交流程,并从法规层面逐个分析了影响进口创新医疗器械同步上市的因素。同时,对比美国食品药品监督管理局(Food and Drug Administration,FDA)的相应程序,分析了同一产品如果在我国和美国同时启动能否达到相近的上市时间,剖析我国与美国法规的差异,并借鉴FDA 控制产品风险的方法,提出了相应政策建议,以期为我国创新医疗器械加速上市提供借鉴与思路。
2.1 创新医疗器械申请
2014 年,《创新医疗器械特别审批程序(试行)》[7] 发布实施。为进一步规范创新医疗器械的申报文件和流程,国家药品监管部门又于2016 年发布了《创新医疗器械特别审批申报资料编写指南》[8]。2018 年,国家药品监管部门对上述2 个文件进行了制修订,并发布了《医疗器械技术审评中心创新医疗器械特别审查申请审查操作规范》[9],目的是更好地扶持创新产品,加快产品上市。
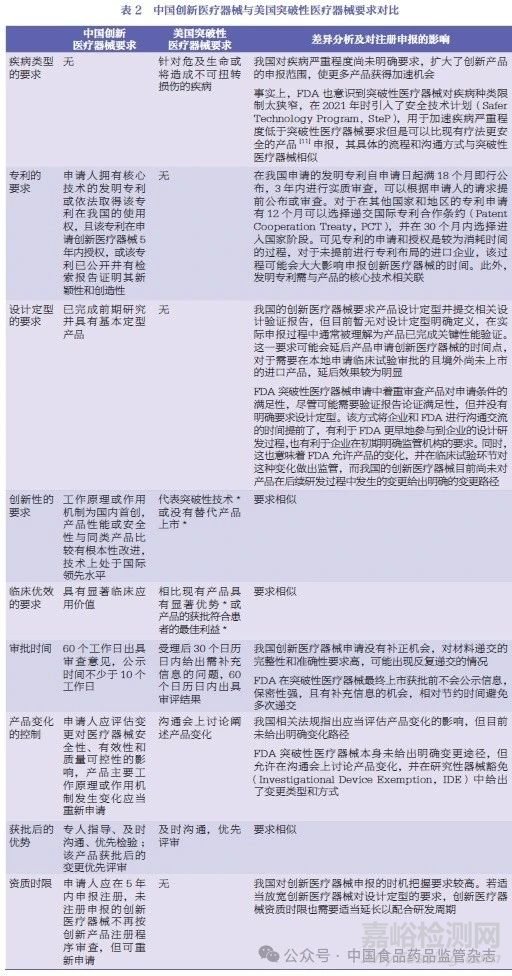
创新医疗器械需要符合以下3 个要求:①产品的创新性。这需要申请人拥有核心技术的发明专利、依法获得该专利的所有权或使用权为证明依据;对于尚未在我国授权的专利,需提供检索报告并以足够的新颖性和创新性证明该专利未来可以获得授权。②该产品应已基本定型,以关键性能的验证报告为证明。③该产品应具有首创性和显著的临床优效性,与同类产品比较时应有根本性的改变,且技术处于领先地位。
FDA 对于突破性医疗器械资质的考察集中在产品的创新性以及临床优效性上。首要条件为该医疗器械为危及生命或将造成不可扭转的损伤的疾病提供了更有效的诊疗手段。另外,突破性医疗器械应满足以下4 个次要条件中的至少1 个:①代表突破性技术;②没有替代产品上市;③相比现有产品具有显著优势;④该产品的获批符合患者的最佳利益[10]。
需要特别说明的是,并不是只有在美国获得了突破性疗法的产品才能在我国获批创新医疗器械,在评估产品是否有替代产品以及与现有疗法相比是否有显著的临床优势时,都是针对本国(地区)市场而言,因此二者并不一定是一一对应的关系。本文将我国创新医疗器械与美国突破性医疗器械的要求进行了对比,总结了法规上的启示并分析了出现上市时间差的原因,见表2。
2.2 临床路径的选择
临床评价路径的选择分为免于临床评价、通过同品种医疗器械评价和通过临床试验数据评价。创新医疗器械具有强创新性,通常不满足免于临床评价的要求。截至2023 年已获批且有公开审评报告的创新产品中,11.2% 的产品采用了同品种的评价方式,82.7% 的产品采用了临床试验,6.1% 的产品采用以上两种混合方式的临床路径完成了产品安全性和有效性的论证。其中,采用临床试验的数据来源既可以是本地临床试验,也可以是境外临床试验。
近年来,为加快医疗器械上市步伐,减轻企业负担,监管部门出台了多项灵活政策,其中包括利用真实世界研究进行临床评价。真实世界研究的数据可以是来自境外已上市国家和地区,用于对我国人种外推或临床条件的补充论证,也可以是来自海南博鳌的特许使用, 用于支持产品的申报注册。目前, 美国艾尔建公司的青光眼引流管XEN Glaucoma Treatment System 和美国强生眼力健公司的“Catalys 白力士”眼科飞秒激光治疗机Catalys Precision Laser System 已成功利用乐城真实世界数据获批。前者从申请到获批仅历时5 个月,后者从首例手术到获批也仅历时14 个月,验证了真实世界数据在临床评价中的可行性,并加速了产品上市进程。根据《关于支持建设博鳌乐城国际医疗旅游先行区的实施方案》[12],博鳌乐城先行区内允许使用“境外已上市但境内未上市”的产品,境外真实世界数据也只能在产品境外上市后获得,因此这两种方式虽然大大缩短了审评审批时间,也尚无法通过这种方式达到同步上市的目标。
我国规定在境内开展临床试验的产品需要提供检测报告用于伦理审批。对于需要临床试验审批的第三类高风险产品,还需在临床备案前获得临床试验批件;未在境外上市的进口产品,需在临床试验审批中提交创新批件。检测报告可以是申请人出具的自检报告或者有资质的医疗器械检验机构出具的检测报告。FDA对开展临床的审批流程与我国类似, 申请人需准备研究性器械豁免(investigational device exemption,IDE)的递交。对于非显著高风险(non-significant risk,NSR) 的产品,IDE 仅需伦理审批;对于显著高风险(significant risk,SR)的产品,还需要FDA 审批IDE 后才能开展临床试验[13]。为此,本文对我国和美国的临床审批流程进行了详细对比,见表3。
2.3 上市递交的要求
完成临床试验并撰写递交所需相关资料后,即可进行上市申请递交。我国与美国创新产品的上市路径及流程类似,审评时限略有不同,具体对比见表4。
2.4 上市流程总结
图3 和图4 总结了我国和美国创新产品上市的流程。从流程图上可以看出,美国的突破性医疗器械资质申请和临床试验申请是两个相对独立的流程,可以同时进行。我国对于无需进行临床试验审批的产品,创新医疗器械的申请和临床试验也可以同步进行,但对于需要临床试验审批且尚未在境外上市的产品,则需要先后进行。根据《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》附件8 中对临床试验审批资料的要求,境外申请人应提交境外上市批件或创新批件[24]。而创新批件的申请又要求产品已定型,也就是说同一款第三类高风险产品如果在我国和美国同时启动,我国在流程上可能会落后2~3 个时间节点。
3、 政策思考
近些年,国家药品监管部门出台了多项政策加速进口医疗器械的上市,包括《接受医疗器械境外临床试验数据技术指导原则》[25]提出可以利用境外数据作为临床评价证据,减少我国境内的临床试验数据规模;《真实世界数据用于医疗器械临床评价技术指导原则(试行)》[26] 规范和引导了利用真实世界研究数据用于医疗器械临床评价,并已在海南博鳌成功试点;新修订《医疗器械监督管理条例》[5] 明确了创新产品可以豁免境外上市批件,进一步提前了进口产品可进行申报的时间点。这些政策有效缩短了我国进口产品与境外上市的时间差。为实现“同步上市”,笔者探索性地提出几点建议,以期进一步缩短进口医疗器械上市时间差。
3.1 适当延长创新医疗器械资格年限,明确临床试验阶段的变更路径,控制产品变更风险
我国申报创新医疗器械时要求产品设计定型,但后续临床环节仍可能发生产品变更,这意味着产品可能需要重新进行部分验证、检测甚至修改临床方案,因此建议可适当放宽创新医疗器械5 年的资格期限,使企业能更充分地进行研发,也避免重复审评创新申请。
同时,为应对申报创新或临床试验备案、审批后发生的变化,建议监管部门应明确变更路径。目前,企业主要的应对方式是与相关部门召开沟通咨询会或者发文咨询,尚没有明确的法规、指南等详细说明变更路径要求。2022 年国家药品监督管理局药品审评中心发布了《药物临床试验期间方案变更技术指导原则(试行)》,其参考了国际上对药品临床阶段发生变更情况的要求,按照变更是否为实质性变更,以及实质性变更是否会影响受试者安全等因素给出了3 条变更路径[27]。因此,建议器审中心也可以参考FDA 的IDE 变更路径,将常见的变更情况分类规范,填补医疗器械临床阶段产品变更的规范的不足,明确每类变更路线所需的资料和审评时间,实现产品的全生命周期监管,引导企业及时沟通、规范申报,避免影响受试者安全,对于加入国际多中心临床试验的企业还可以更好地协调境外临床的时间。
3.2 统一临床试验审批中对境内和境外申请人的要求,取消进口产品提交上市证明或创新医疗器械批件的要求
《需进行临床试验审批的第三类医疗器械目录(2020 年修订版)》中,对应进行临床试验审批对象的要求为“与境内外已上市产品相比,采用全新设计、材料或机理,和/ 或适用于全新适用范围”[14],也就是说,需要申报临床试验审批的产品在某一方面是没有可替代已获批产品的,即便并非为创新医疗器械,也可以填补市场空白。在该背景下,建议可适当考虑优先患者利益,及时将新产品引入市场。尽管相关政策为我国企业提供了一定的政策倾斜,但是临床试验审批的目的是考察产品进入临床试验的安全性。因此,笔者认为只要进口产品可以提供证明临床风险合理可控的证据,并非必须通过境外上市论证临床安全性才可以开展临床试验。同时,进口医疗器械的快速引入也有利于促进国内企业的研发和创新。
3.3 促进国际标准本地转化和更新,统一国内外审评尺度
《国家药品监督管理局 国家标准化管理委员会关于进一步促进医疗器械标准化工作高质量发展的意见》提出:“建立与国际标准快速联动的标准更新机制,探索国内标准与国际标准同步立项,缩短国际标准转化周期。”[28] 目前,我国医疗器械国际标准转化率已达90% 以上,其中最新版的转化标准占全部的53%[29]。及时转化国际标准,可以避免同一产品按照不同的标准和评价体系试验,对促进医疗器械贸易和国际化监管都有积极作用。此外,政策的差异带来的影响是双向的,即进口创新产品难以在我国同步上市,我国出口的高价值医疗器械又相对较少。除了需要促进提高我国研发能力以外,与国际靠拢的审评政策调整也有利于为我国医疗器械走出国门做铺垫,以引导我国产品与国际标准接轨。
3.4 完善专利制度,树立可信可靠国家形象
实现进口创新器械在境内外的同步上市,需要在产品研发早期就在我国申报专利,向审评机构递交未公开的研发细节文件,并将尚未完全曝光的样品送到相关部门检测,这些都需要我国在国际上建立可信可靠的形象,取得进口企业的信任,具体措施包括:完善专利及知识产权管理保护工作,将专利保护制度落实到研发、检测、临床、上市审批及上市后监管全过程,充分保护创新成果。同时鼓励我国创新医疗器械企业尽早进行全球专利布局,为日后产品走出国门做准备。
3.5 允许创新产品自主灵活定价,为全球统一定价提供空间
2022 年9 月, 在《国家医疗保障局对十三届全国人大五次会议第4955 号建议的答复》中,答复了关于将创新产品纳入医保绿色支付通道的建议,并指出:由于创新医疗器械临床使用尚未成熟、使用量暂时难以预估,尚难以实施带量方式。在集中带量采购之外留出一定市场为创新产品开拓市场提供空间[30]。无论是国产还是进口企业,创新医疗器械都面临着研发周期长、投入大、医师培训任务重、市场推广难以及回收成本时间漫长的问题,暂时不纳入集采有利于给创新企业留出合理的利润空间,发挥市场经济的作用促进发展,对于进口企业也可以更灵活地实现全球统一定价管理。
3.6 鼓励境内企业以多种形式开展海外合作并以国产申请人身份注册申报
根据《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》附件8 中的要求,境内申请人应提交企业营业执照或事业单位法人证书,而境外申请人应提交企业资质证明文件及境外上市批件或创新批件。由于临床试验审批中对进口医疗器械和国产医疗器械的要求不同,将可能影响进口产品的全球同步研发,难以把我国列入国际多中心临床试验。但若进口企业与我国相关企业进行合作并以境内申请人的身份申报,则可以更早地进入临床阶段。因此,以多种方式参与海外创新活动也是促进国际技术交流、提升我国研发能力、带动上下游产业发展的重要途径,包括联合开发、合并收购以及委托生产等。例如,沛嘉医疗科技(苏州)有限公司与法国HighLifeSAS 公司签署了合作协议,获得了法国HighLife SAS 公司经导管二尖瓣置换术有关产品的独家许可,将负责该产品在我国的制造、开发和商业化。作为需要临床试验审批的第三类高风险产品,沛嘉医疗科技(苏州)有限公司利用这种合作模式将进口技术以境内申请人的身份引入我国,加快了该产品的上市进程。
4、结语
自创新医疗器械注册程序实施以来,我国通过该途径申报和获批的产品逐年增加,我国各级部门包括医疗器械监管部门、检测机构、临床试验机构均出台了相应政策,缩短了审评审批时长,推动创新医疗器械的加速上市,但尚未达到与境外同步上市的目标。为进一步实现进口创新医疗器械在我国同步上市的目标,本文提出了相关政策建议,以促进进口创新医疗器械在我国同步上市。相关建议不仅使进口企业受益,吸引全球资本,也有利于促进我国企业与国际标准接轨,提升专利布局意识,加强国际合作,不断缩短与国际先进水平的差距,实现以人民健康为中心的发展目标。
参考文献
[1] 国家发展和改革委员会. 中华人民共和国国民经济和社会发展第十四个五年规划和2035 年远景目标纲要[EB/OL]. (2021-03-13) .https://
www.gov.cn/xinwen/2021-03/13/content_5592681.htm#:~:text=%E4%B8%AD%E5%8D%8E%E4%BA%BA%E6%B0%91%E5%85%B1%E5%92%8C%E5%9B%BD%E5%9B%BD
,%E5%85%B1%E5%90%8C%E7%9A%84%E8%A1%8C%E5%8A%A8%E7%BA%B2%E9%A2%86%E3%80%82.
[2] 工业和信息化部 国家发展和改革委员会 科学技术部 商务部 国家卫生健康委员会 应急管理部 国家医疗保障局 国家药品监督管理局
国家中医药管理局关于印发“十四五”医药工业发展规划的通知 [EB/OL]. (2022-01-31) . https://www.gov.cn/zhengce/zhengceku/2022-01/31/content_5671480.htm.
[3] 国家药品监督管理局,国家发展和改革委员会,科学技术部,等.“十四五”国家药品安全及促进高质量发展规划 [EB/OL].(2021-12-30).
https://www.nmpa.gov.cn/directory/web/nmpa///yaopin/ypjgdt/20211230145247117.html.
[4] 国家市场监督管理总局. 医疗器械注册与备案管理办法[EB/OL].(2021-08-26) .https://www.samr.gov.cn/zw/zfxxgk/fdzdgknr/fgs/art/2023/art_568880e3ee344c45b38d073bba1c53ad.html.
[5] 国务院. 医疗器械监督管理条例[EB/OL]. (2021-03-19). https://www.gov.cn/zhengce/content/2021-03/18/content_5593739.htm.
[6] 王兰明, 赵阳. 深化医疗器械审评审批制度改革, 促进医疗器械产业高质量发展:中国医疗器械审评审批制度改革概述[J]. 中国食品药品监管,2021(1):16-28.
[7] 国家食品药品监督管理总局. 关于印发创新医疗器械特别审批程序( 试行) 的通知[EB/OL].(2014-02-07) .https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/fgwj/gzwj/gzwjylqx/20140207154501788.html.
[8] 国家药品监督管理局. 关于发布创新医疗器械特别审查申报资料编写指南的通告(2018 年第127 号)[EB/OL].(2018-12-12) .https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20181105160001106.html.
[9] 国家药品监督管理局医疗器械技术审评中心. 关于发布医疗器械技术审评中心创新医疗器械特别审查申请审查操作规范的通告(2018 年第11 号)[EB/OL]. (2018-11-29).https://www.cmde.org.cn//xwdt/shpgzgg/gztg/20181129151100780.html.
[10] FDA. Breakthrough Devices Program[EB/OL]. (2023-02-24).https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program.
[11] FDA. Safer Technologies Program (SteP) for Medical Devices[EB/OL]. (2021-03-18).https://www.fda.gov/medical-devices/how-study-andmarket-your-device/safer-technologies-program-step-medical-devices.
[12] 国家发展改革委,国家卫生健康委,国家中医药局,等. 关于支持建设博鳌乐城国际医疗旅游先行区的实施方案[EB/OL].(2019-09-10).
https://www.gov.cn/xinwen/2019-09/17/content_5430452.htm.
[13] FDA. Investigational Device Exemption (IDE)[EB/OL]. (2022-10-03). https://www.fda.gov/medical-devices/premarket-submissionsselecting-and-preparing-correct-submission/investigational-device-exemption-ide.
[14] 国家药品监督管理局. 国家药监局关于发布需进行临床试验审批的第三类医疗器械目录(2020 年修订版)的通告(2020 年第61 号)[EB/OL].(2020-09-18). https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20200918103742111.html.
[15] The Federal Register. 42 CFR Part 11 [EB/OL].(2016-09-21). https://www.ecfr.gov/current/title-42/chapter-I/subchapter-A/part-11.
[16] FDA. Changes or Modifications During the Conduct of a Clinical Investigation; Final Guidance for Industry and CDRH Staff [EB/OL].(2001-05-29). https://www.fda.gov/media/72429/download.
[17] The Federal Register. 21 CFR 820. Quality System Regulation [EB/OL]. (2023-03-01). https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820.
[18] The Federal Register. 21 CFR 58 Good Laboratory Practice for Nonclinical Laboratory Studies [EB/OL]. (2023-12-22). https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-58.
[19] 国家药品监督管理局. 国家药监局关于发布《医疗器械注册自检管理规定》的公告(2021 年第126 号)[EB/OL]. (2021-10-22).https://www.nmpa.gov.cn/xxgk/fgwj/xzhgfxwj/20211022153823130.html.
[20] 王保亭,耿鸿武. 医疗器械蓝皮书:中国医疗器械行业发展报告(2022)[M]. 北京: 社会科学文献出版社,2022.
[21] FDA. FDA and Industry Actions on Premarket Notification (510(k)) Submissions: Effect on FDA Review Clock and Goals [EB/OL]. (2022-10-03).www.fda.gov/media/73507/download.
[22] FDA. FDA and Industry Actions on Premarket Approval Applications (PMAs): Effect on FDA Review Clock and Goals [EB/OL]. (2022-10-03).
www.fda.gov/media/73504/download.
[23] FDA. FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review Clock and Goals [EB/OL]. (2022-10-03).www.fda.gov/media/107652/download.
[24] 国家药品监督管理局. 关于公布医疗器械注册申报资料要求和批准证明文件格式的公告(2021 年第121 号)[EB/OL].(2021-09-30).
https://www.nmpa.gov.cn/ylqx/ylqxggtg/20210930155134148.html.
[25] 国家食品药品监督管理总局. 总局关于发布接受医疗器械境外临床试验数据技术指导原则的通告(2018 年第13 号)[EB/OL].(2018-01-11).
https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20180111175801772.html.
[26] 国家药品监督管理局. 关于发布真实世界数据用于医疗器械临床评价技术指导原则(试行)的通告[EB/OL].(2020-11-26) .https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20201126090030150.html.
[27] 国家药品监督管理局药品审评中心. 国家药监局药审中心关于发布《药物临床试验期间方案变更技术指导原则(试行)》的通告(2022 年第34 号)[EB/OL].(2022-06-23). https://www.cde.org.cn/main/news/viewInfoCommon/c9d649a44ba90b52ceb8072c28da768f.
[28] 国家药品监督管理局,国家标准化管理委员会. 关于进一步促进医疗器械标准化工作高质量发展的意见[EB/OL]. (2021-03-30). https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjylqx/20210330170905141.html.
[29] 许慧雯, 孟芸, 邵姝姝, 等. 监管法规协调背景下医疗器械国际标准转化研究与思考[J]. 中国药事,2022,36(12):1350-1357.