您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-05-21 08:10
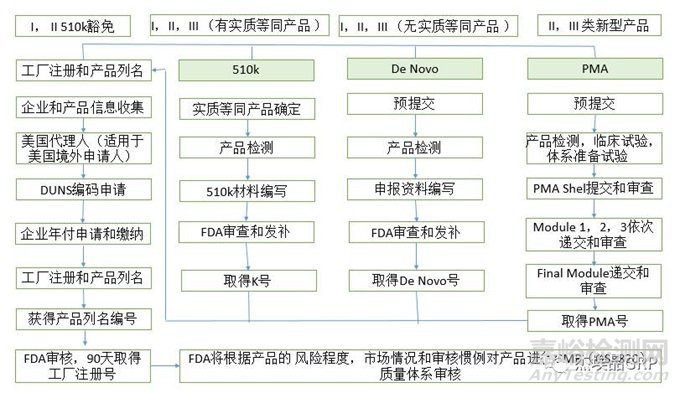
医疗器械美国FDA认证注册流程图
FDA是美国食品与药品监督局(US Food andDrug Administration)的缩写。在美国市场销售的食品,药品,化妆品,医疗器械,烟草均由FDA监管。
FDA对医疗器械的管理通过器械与放射健康中心(CDRH)进行。
FDA对医疗器械实行分类管理,根据风险等级和管理程度把医疗器械分成三类(Ⅰ,Ⅱ,Ⅲ)进行上市前管理,Ⅲ类风险等级最高。FDA将每一种医疗器械都明确规定其产品分类和管理要求,任何一种医疗器械想要进入美国市场,必须首先弄清申请上市产品分类和管理要求。
FDA从科学、工程和临床专家以及消费者和工业组织推荐的候选人中挑选出一些专家组成分类专家委员会,其中消费者和工业组织代表没有投票权。FDA会根据专家委员会的建议最终决定医疗器械产品的详细分类,在定期公布这些分类结果的同时,每年还会对法规代码库进行更新。
在明确了以上信息后,企业就可以着手准备有关的申报资料,并按一定程序向FDA申报以获取医疗器械FDA认证批准。对于任何产品,企业都需进行企业注册(Registration)和产品列名(Listing)。
I类产品为“普通管理(General Controls)”产品,是指风险小或无风险的产品,如医用手套、压舌板、手动手术器械等,这类产品约占全部医疗器械的30%。FDA对这些产品大多豁免上市前通告程序,一般生产企业向FDA提交证明其符合GMP并进行登记后,产品即可上市销售。
II类产品为“普通+特殊管理(General & Special Controls)”产品,其管理是在“普通管理”的基础上,还要通过实施标准管理或特殊管理,以保证质量和安全有效性的产品,这类产品约占全部医疗器械的62%。FDA只对少量的II类产品豁免上市前通告程序,其余大多数产品均要求进行上市前通告(510K)。生产企业须在产品上市前90天向FDA提出申请,通过510K审查后,产品才能够上市销售。
III类产品为“上市前批准管理(Pre-market Approval,PMA)”产品,是指具有较高风险或危害性,或是支持或维护生命的产品,例如人工心脏瓣膜、心脏起搏器、人工晶体、人工血管等,这类产品约占全部医疗器械的8%。FDA对此类产品采用上市前批准制度,生产企业在产品上市前必须向FDA提交PMA申请书及相关资料,证明产品质量符合要求,在临床使用中安全有效。FDA在收到PMA申请后45天内通知生产企业是否对此申请立案审查,并在180天(不包括生产企业重新补充资料的时间)内对接受的申请做出是否批准的决定,只有当FDA做出批准申请的决定后,该产品才能上市销售。
总体来讲,FDA总部对近60%的医疗器械进行上市前通告(510K)或者上市前批准(PMA)的审查。

来源:Internet


