您当前的位置:检测资讯 > 生产品管
嘉峪检测网 2024-07-01 08:36
在医疗器械的质量管理体系框架内,CAPA(Corrective and Preventive Actions)扮演着重要角色。它不仅是一种关键的管理工具,还是提升和维持产品质量的有效策略。
与此同时,药监局在进行审核时,也将CAPA作为评估的重点内容之一。
本文融合现行法规与检查要求,对CAPA进行简明扼要的探讨。
No.1 CAPA的相关法规
1). ISO13485和GB/T42061的要求
ISO13485和GB/T42061中8.5.2和8.5.3条款规定:企业应采取措施消除不合格原因和潜在原因,以防止不合格品再次发生。
2). QSR 820的要求
美国QSR 820中第100条款规定:每个制造商应建立和保持实施纠正和预防措施的程序。
3). 医疗器械生产监督管理办法
《医疗器械生产监督管理办法》第三十七条规定:医疗器械企业应当建立纠正/预防措施和程序,确定产生问题的原因和根本原因,采取有效措施,防止相关问题再次发生。
No.2 CAPA的术语与定义
1). 纠正(Correction)
为消除发现的不合格品或行为所采取的措施。
2). 纠正措施(Corrective Action)
为消除不合格的原因并防止再发生所采取的措施。
3). 预防措施(Preventive Action)
为消除潜在不合格或者其他潜在不期望情况的原因所采取的措施。
便于理解,举例说明:
不合格描述:产品尺寸不符合要求。
纠正:产品报废处理;或返工,直至符合要求。
纠正措施:调整相关加工工艺或工装,直至产出合格产品,防止该不合格产品再次发生。
预防措施:调整其他类似的所有加工工艺,防止类似问题再次发生。
No.3 CAPA的体系文件
1). 纠正预防措施控制程序
公司应按照ISO13485或QSR 820的要求,建立《纠正措施控制程序》和《预防措施控制程序》,并保持其有效运行。
ISO13485中“纠正”和“预防”是分成两个条款的,而QSR 820中“纠正”和“预防”是合并在一起的。
所以,也可以合并成一个文件《纠正预防措施控制程序》,形式上没有具体的规定。
2). 数据分析程序
公司应使用适宜的统计学方法或工具收集并分析不合格品的数据。
可以是外部的数据,如:客户投诉、药监局检查、客户审查等。
也可以是内部的数据:如:不合格品报告(NCMR)、内审或管理评审、过程控制数据(SPC)等。
No.4 CAPA的基本流程
CAPA流程或表格的形式上可能有所差别,但至少都应该包括如下内容:
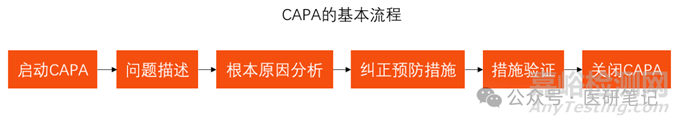
1). CAPA的启动
根据公司程序文件的规定,确定启动相应的CAPA。
2). 问题的描述
问题的描述应该包括:
问题的是什么(What)?
什么时候发生的(When)?
在哪发生的(Where)?
谁发现的(Who)?
受影响的范围或程度是什么(How much)?
总之,问题描述越详细越好,必要时可以提供图片、往来邮件、单据等作为证据。
但是,不能有主观的猜测或结论,只描述客观事实。
3). 根本原因分析
根本原因(Root Cause)有的比较容易发现。
有的隐藏得比较深,需要组织团队进行调查分析。
有的问题是由多种原因导致的。
必要时,可以借助于适当的分析方法或工具,如:
5个为什么分析(5 Why),多问几个为什么,刨根问底,尽可能往深了挖。
鱼骨图:可以从人、机、料、法、环、测等方面逐一分析。
故障树:利用逻辑因果关系,从上往下逐层剖析。
排除法:这条在实际中应用也比较多,对于无法准确确定原因的情况,通过改变某一个变量,进行逐一排除,直至找到精准的原因。
当然还有很多其他的工具,大家结合各自的实际情况及喜好,选择适合自己的工具。
4). 纠正预防措施
根本原因找到后,应该制定相应的纠正预防计划,需要落实到具体负责人,制定具体完成的时间进度等。
计划制定好后,应该按照计划逐步实施,必要时,公司应配备足够的资源支持。
必要时,措施中还应该对以前的批次进行追溯和评估。
5). 纠正预防措施的验证或确认
纠正预防措施应该得到验证或确认,应评估:
这些措施能否消除根本原因;
这些措施能否涵盖所有可能受影响的产品或流程;
这些措施是否会产生新的不利影响;
这些措施是否会带来新的风险等。
经过确认后的措施,应该进行固化,形成标准化流程,对相关人员进行培训。
6). CAPA的关闭
措施经过验证确认有效后,收集汇总数据。
然后提交小组评审,由管理者代表或其他指定管理人员签字确认,关闭CAPA。
最后,别忘记了随时更新CAPA清单。

来源:医研笔记


