您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-07-17 08:27
对我国与美国、欧盟、日本为代表的其他 ICH主要国家和地区的现行法规和当前监管实践的对比,提炼在药品注册检验方面的差异,以供我国持续调整和改进药品注册检验 管理的参考借鉴。为了突出相关信息和因素,以表格形式归纳如下。
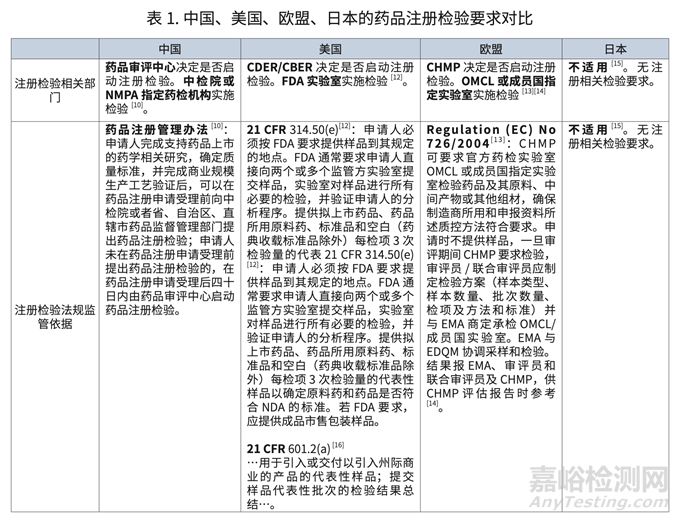

对比所示,我国长期以来将药品注册检验的实验研究过程作为核准药品注册标准的检验技术支撑,在当前监管实践中,进口药品标准复核研究工作具有为口岸药品检验提供科学实验基础,使制药企业遵循我国的监管要求的历史意义。
但其他 ICH 国家和地区并不将药品注册检验作为常规的注册管理要求,仅在涉及批签发或者有因的特别情形下在注册审评过程中要求检验,注册标准的核定工作也主要基于对申报资料的技术审评,而非对样品的实验室检验。
另一方面,我国的药品上市后监管以药品注册标准为判定假药、劣药的依据,假药、劣药 的处罚决定应当依法载明药品检验机构的质量检验结论,官方药检机构的检验是我国药品上市后监管的重要措施。因此,在药品注册审评过程中会格外关注核准的药品注册标准在上市后官方药检机构开展监管检验的实验室适用性。可见,在讨论其他 ICH 国家和地区药监实践对药品注册检验管理的调整改进的借鉴意义时,也需进一步研究其上市后药品检验的要求。如下表归纳:
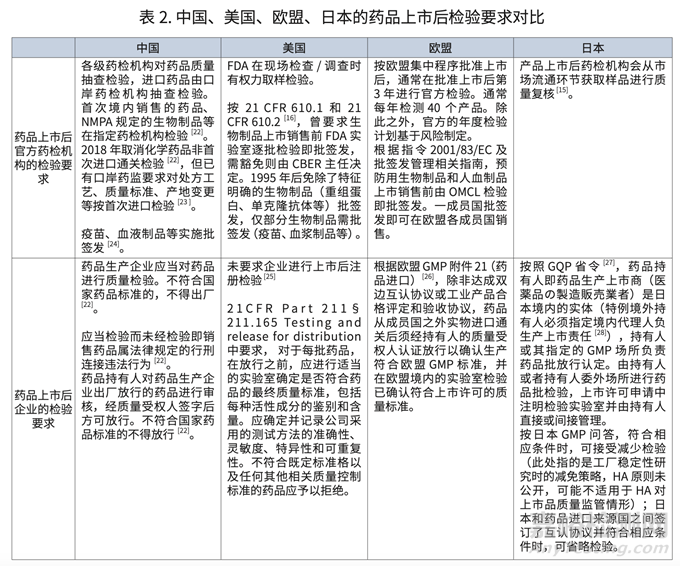
我国投入巨大的行政和技术资源、由官方药检机构开展上市药品的质量抽查检验。相较之下,除了批签发管理的疫苗、血液制品等药品外,其他 ICH 国家和地区则更偏向于要求持有人履行责任,确保药品质量并上市药品的放行检验。
国外经验对我国药品注册检验管理的启示
通过前述药品注册检验要求方面中国与 ICH 其他国家对比的异同,值得借鉴的启示如下:
在临床试验申请阶段,我国与美国、欧洲和日本基本一致,临床试验申请审评过程中均不要求开展药品注册检验(疫苗等特殊情形除外),否则难以实现较快完成临床试验的审评审批。我国自 1978 年开始要求药物临床试验申请注册检验,到 2007 年免除了除生物 制品外的药物临床试验申请的注册检验,再到 2018 年免除了除疫苗外的所有药物临床试验申请的注册检验,是一个基于风险考虑逐步探索、逐步放开的过程。这个过程既是药品监管科学的发展,也与我国制药行业的整体合规水平以及监管合规整顿成果相关。
行业更有序的发展现状能够支撑基于风险考虑采取的更高效的监管措施,取消注册检验, 显著地加快了临床试验申请的审评审批速度(从 2017 年首轮审评审批平均用时约为 120 个工作日,到当前 60 工作日内完成审评审批),有力促进了新药创制和中国参与新药全球同步 研发,更好地满足了患者用药需求。
• 在上市许可申请阶段,1978 年至今,上市许可申请审评时我国一直将药品注册检验尤其是标准复核的实验研究作为核定药品注册标准的技术依据 。虽然通过工作程序的调整和优化,将注册审评和注册检验由依次串行调整为大致的同时并行开展,并且允许将注册检验前置于上市许可申请提交前开展,但囿于实验研究客观上的耗时周期、尤其是新分析方法转移时不可避免的实验室适用性探索过程,药品注册检验仍是制约上市许可审评审批提速的关键步骤。与之相较,美国、欧洲和日本的监管实践具有借鉴意义:
1)· 美国· 尽管法律规定了 FDA 有权启动注册检验且申请人应留样备检,但除了审评阶段即启动批签发程序的生物制品外,FDA 在上市许可审评过程中鲜少实际启动检验。 FDA 基于风险通常不启动上市许可申请注册检验、如有因情形可在审评过程中的现场检查时要求现场检验或者抽样送往 FDA 实验室检验、以及在上市许可申请审评期间 即通过FDA 实验室检验开展批签发相关工作的做法,值得我国借鉴。
2) 欧盟 与美国相似,欧盟虽有药品注册检验的法律依据,但实践中注册检验并非常规措施而是有因触发的特别措施。审评中发现问题启动检验与审评中发现问题启动检查的严重程度相当,并且一旦基于审评要求启动注册检验,审评员需制定具体的检验要求和方案,值得我国借鉴。
我国已有关于注册审评期间基于风险、基于审评需要启动检验和核查的规范性文件,但目前仅对启动核查的风险因素和风险等级给予较为明确的考虑,检验涉及的品种风险因素和等级判断,且研发生产主体合规因素和风险等级管理的指导原则尚在征求意见,尚缺乏针对启动注 册检验的风险因素和风险等级判定的细则。有必要参考欧盟的做法,基于明确的审评考虑启动注册检验,且一旦启动注册检验,应当针对审评需要的具体检验问题制定具体的检验要求和方案。
3) 日本 日本上市许可申请注册程序中并未设置注册检验的流程,日本要求药品批准上市后持有人必须在日本境内的实验室(持有人的自有实验室或者委外实验室)完成批 检验放行,持有人有责任管理批检验放行的实验室,且在上市许可申请中注明该实验室的做法,值得我国借鉴。
如前所述,美国、欧盟和日本均不对上市后变更启动注册检验。基于风险的考虑和开展注 册检验的目的,建议减少启动注册检验的上市后变更事项,尤其是对于不改变药品注册标准的 检项及检验方法、未产生超出现有药品注册标准控制范围的新的关键质量属性的药学变更(工艺变更、原辅包供应来源变更、生产场地变更等)可考虑不启动药品注册检验。
对于已有药品注册标准发生改变必须对其科学合理性重新复核、或者已有药品注册标准不足以检出变更产生的新的关键质量属性变化的情形,若确需通过药品注册检验的实验研究支持药品审评,则建议基于审评需要提出针对性的注册检验要求,制定结构化的检验要求细则以便持有人能够据此预见可能需要启动标准复核的检项和内容,并能够据此在提交变更补充申请前 提出前置的注册检验,减少注册检验对审评审批时限的影响。
此外,除了上述质控技术因素外,还建议将药品研发生产主体的合规因素纳入启动药品注 册检验、尤其是上市后变更启动药品注册检验的考虑,审评过程中基于企业的合规情况建立信用记录,对合规情况良好的企业减少药品注册检验。
• 在药品上市流通过程中,我国更加依赖各级药检机构进行上市药品质量抽查检验和口岸药检机构进行药品进口通关抽查检验。相较而言,美国、欧盟和日本更强调药品持有人的责任,包括由持有人完成上市销售药品的检验以确保药品质量。
参考文献:药品注册检验管理相关问题研究, RDPAC,2023.12

来源:文亮频道


