您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-08-12 08:50
对于境外已上市境内未上市的药品,需结合药品具体情况,按照临床评价的基本逻辑对原研药品的临床研究数据进行充分评价,根据评价结果确定临床试验路径。对于不同研发背景的药品,其所需开展的临床试验应具体问题具体分析。
1、法规要求
根据化学药品注册分类改革工作方案(2016年51号),化药3类指境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。
原研药品指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。
根据《境外已上市境内未上市药品临床技术要求》(2020年29号),境外已上市境内未上市药品的临床技术要求,应遵循临床评价基本逻辑,在充分评价中国患者临床需求、境外原研药品临床安全性和有效性、以及种族因素影响的基础上,基于中国患者获益/风险评估的需要,确定其在境内上市需开展的临床试验技术要求。
临床评价基本逻辑
(一)临床需求评估
对中国患者临床需求的程度做出判断。
(二)有效性和安全性评价
(三)种族敏感性分析
(四)基于中国患者获益/风险评估进行决策
CDE培训会议中也讲到通常豁免临床是比较难的,通常也只是限于严重危及生命及罕见病等的一些优先审评品种。

2、案例分析
根据实际情况化药3类又可以分为真3类(全新的化合物进入国内)、半真3类(国内有其它剂型产品)、假3类(国内有同品种仿制药上市多年),不同情况所进行的临床试验也不同。
3类化药可能需要进行的临床试验情形如下:
第一种是豁免BE及其他临床试验,由境内外临床数据支持,通过体外评价证实与原研一致;
第二种是BE试验,可豁免验证性临床试验仅以BE试验申请注册;
第三种是验证性临床试验,无法或无需进行BE试验的品种,通过验证性临床试验证实有效及安全性;
第四种是BE+验证性临床试验,BE证实与原研生物等效,验证性临床确证疗效及安全性;
第五种是剂量探索(I期)+确证性临床研究,有种族差异的,需以新药临床思路进行仿制药临床研究。
根据BE试验中的PK数据,可以结合原研公布的PK数据进行对比,多角度分析其是否具有PK方面的种族差异,若无PK差异,则可考虑直接开展验证性临床;若有种族差异,按照临床评价的基本逻辑则还应考察中国人群的剂量探索性试验,为验证性临床提供剂量依据。
(1)BE+验证性临床
做了验证性临床的3类仿制药绝大多数都是国内首次获批的化合物,如奥贝胆酸片,江苏恒瑞只做了BE进行申报未被批准;正大天晴做完BE 后,后面申请了3期临床。
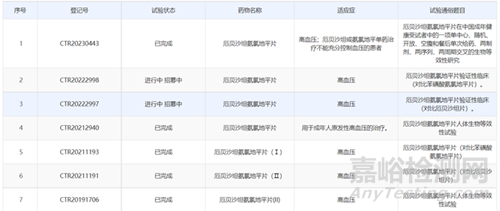
氟比洛芬酯注射液(特殊注射液),该品种虽然有2家仿制药上市,但原研未进入国内,仍按3类申报,武汉大安进行了3期临床、1期临床和PK试验;广东嘉博制药有限公司2018年完成BE试验后进行申报未被批准,2020年完成III期临床后进行申报,目前已经获批。四川科伦、山东威高、扬子江药业等没做临床的企业全部被毙掉,根据审评结果可以发现,仿制该品种仍然需要进行有效性和安全性的临床试验:像氟比洛芬酯这种情况的项目还有很多品种。
同类药物CDE临床登记情况如下:
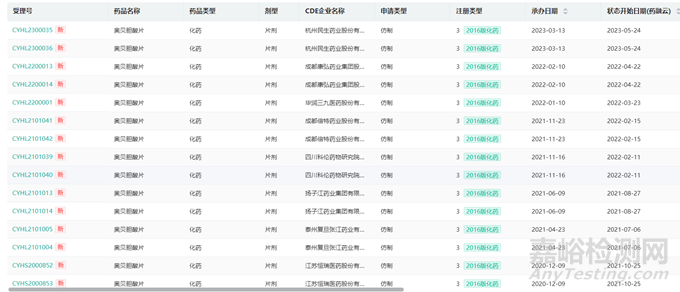
|
|
药物名称 |
临床试验类型 |
|
1. |
替米沙坦氨氯地平片 |
参比制剂原研已经进口,生物等效性试验 |
|
2. |
奥美沙坦酯氨氯地平片 |
参比制剂原研地产化品种,生物等效性试验 |
|
3. |
缬沙坦氨氯地平片(Ⅱ) |
参比制剂原研已经进口,生物等效性试验 |
|
4. |
阿齐沙坦氨氯地平片 |
参比制剂未进口原研药品,日本橙皮书,CDE临床登记情况如下,BE+验证性临床
  |
|
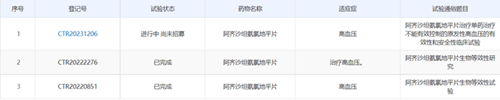
5 |
厄贝沙坦氨氯地平片 |
参比制剂未进口原研,欧盟、日本上市。
根据CDE临床登记为BE加验证性临床试验
|
(2)生物等效性试验(BE)
1)仅进行BE获批的品种中,此类品种多为缓控释制剂,例如盐酸美金刚缓释胶囊、苯扎贝特缓释片等,由于其普通片剂已在国内上市多年,因此仅需BE证实与原研生物等效,豁免验证性临床试验。

特例,拉考沙胺注射液国内首仿企豁免临床的关键在于该药的药代特性为口服生物利用度约为100%,原研片剂批准进口,有国内临床数据充分,注射剂通过与片剂BE等效豁免临床。
(3)豁免临床
厂家没做验证性临床也获批上市的注射剂产品有:盐酸溴己新注射液、克林霉素磷酸酯注射液、硫酸特布他林注射液、左西孟旦注射液和注射用右雷佐生等。这些不需要做临床的产品都有一个特点,就是仿制药厂家已经在国内上市多年的注射剂。
原研小针及国产粉针、小针上市多年,疗效安全性充分验证;100ml:4mg唑来膦酸注射液的原研产品明确,与已上市品种适应症、用法用量相同,最终获准豁免验证性临床试验。
3、临床试验技术要求
已废止的旧版《药品注册管理办法》2007年28号以及对应的附件2:化学药品注册分类及申报资料要求
“3.已在国外上市销售但尚未在国内上市销售的药品:(1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂;”
“2.属注册分类3和4的,应当进行人体药代动力学研究和至少100对随机对照临床试验。多个适应症的,每个主要适应症的病例数不少于60对。避孕药应当进行人体药代动力学研究和至少500例12个月经周期的开放试验。
属于下列二种情况的,可以免予进行人体药代动力学研究:
(1)局部用药,且仅发挥局部治疗作用的制剂;
(2)不吸收的口服制剂。”
这是之前3.1类(即现在的新3类)需要做临床的法规依据。
现行《药品注册管理办法》2020年27号以及对应的NMPA 化学药品注册分类及申报资料要求 20200629
“3类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与参比制剂的质量和疗效一致。”属于之前3.1类,但是去掉了明确的临床试验要求。而是在“管理办法”正文中:
“第三十五条 仿制药、按照药品管理的体外诊断试剂以及其他符合条件的情形,经申请人评估,认为无需或者不能开展药物临床试验,符合豁免药物临床试验条件的,申请人可以直接提出药品上市许可申请。豁免药物临床试验的技术指导原则和有关具体要求,由药品审评中心制定公布。”后面CDE出台了《境外已上市境内未上市药品临床技术要求》(2020年29号)
新的药品注册管理办法对3类药取消了“PK+100例”随机对照临床试验的规定,制定出以科学为基础、同时基于ICH E5、E17、E6技术要求,更加灵活科学的临床试验方案。新3类取消了监测期,对首仿和后续申报申请人的临床研究要求尚未在公开的规定上予以明确,3类药开展验证性临床试验存在一定的顾虑。
CDE后续是否可行考虑首家获批后给予市场独占期,之后申报者可以BE或者直接报产,这样给首家带来好处的同时也避免了资源浪费,无需重复做临床试验。
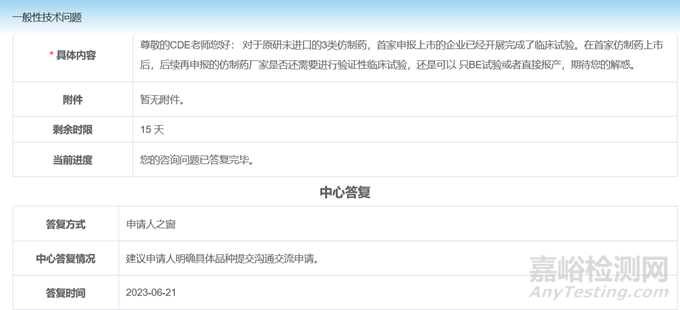
此外,从网络各个渠道能得到的CDE审评人员对于该问题的回复,基本上是“企业需要自行评估,case by case”以及“基于充分的数据与分析,与CDE提前沟通。
通过CDE一般性技术问题咨询得到的答复也是建议申请人明确具体品种提交沟通交流申请。

来源:注册圈