您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-01-09 09:05
摘要
相比化学仿制药,化学创新药由于其结构新颖,降解途径未知等特点,在强制降解研究的主要降解产物归属,降解试验条件摸索,降解产物的致突变性研究等方面需更多尝试。目前,已有多篇针对药物强制降解研究的文献综述,但未对化学创新药的强制降解研究进行针对性的论述。本文参考国内外相关文献,并结合在药物研发中的实践,重点对化学创新药强制降解研究的主要降解产物归属,降解试验条件摸索及降解产物的致突变性等内容进行讨论,以期为药物研发人员提供一些借鉴。
关键词:化学创新药;强制降解;一般考虑;降解试验条件;降解产物;致突变性
化学药物的强制降解研究在药物研发过程中扮演着十分重要的角色,其具体作用可能包括但不限于:(1)有助于确定分子的内在稳定性,鉴定药物可能的降解产物,确定降解路线揭示降解机理;(2)验证所用的分析方法对药物稳定性的指示能力;(3)指导药品处方工艺和包装系统的开发,确定药品贮藏条件;(4)对药物的安全性和毒性进行评估;(5)指导药物的盐基和晶型的选择;另外,强制降解试验会产生药物的代谢产物,有助于加强对药物吸收、分布、代谢与排泄的理解[1]。
目前,已有多篇针对药物强制降解研究的文献综述[2-7],但未对化学创新药的强制降解研究进行针对性的论述。相比化学仿制药,化学创新药由于其结构新颖、降解途径未知等特点,在强制降解研究的主要降解产物归属、降解试验条件摸索、降解产物的致突变性研究等方面需更多尝试。本文参考国内外相关文献,并结合在药物研发中的实践,针对化学创新药强制降解研究进行讨论,以期为药物研发人员提供一些借鉴。
1、创新药降解反应条件的选择
药物的强制降解研究通常包括酸、碱、氧化、高温和光照降解 5 个方面。一般认为,强制降解条件应结合药物的性质制定,所采用的强制降解条件(或降解反应的终点)应使药物所发生的降解途径接近药品在真实情景(长期试验)下所产生的降解。过度降解(或过于剧烈的降解条件)会形成人为或伪降解产物或次级降解产物而失去强制降解研究的意义,同时也给药物的稳定性分析方法的开发带来不必要的工作量。无论是仿制药还是创新药,其强制降解均应按上述原则进行研究。
已有文献进行仿制药强制降解研究的报道,如左氧氟沙星注射液的日本药品医疗器械管理局(Phamaceuticals and Medical Deviees Ageney PMDA)审评文件中显示了左氧氟沙星原料药的强制降解条件,有文献报道头孢曲松钠在酸(3% 醋酸,室温30min) 、碱(0.1mol·L-1碳酸氢钠,60℃下 30min) 降 解 条 件 下 可 得 到 数 量 适 中 的 杂 质等[8-10]。相比仿制药,创新药的强制降解研究具有阶段性和摸索性的特点,美国FDA鼓励在新药Ⅰ、Ⅱ期临床试验阶段进行强制降解研究,要求在Ⅲ期临床 阶 段 以 及 新 药 申 请 (new drug application,NDA)阶段应进行强制降解研究,并以年报的方式进行提交[11]。创新药的光照强制降解试验可参照ICHQ1B 中的光照稳定性试验条件进行,即可见光1.2×106Ix·h,近紫外200W·h·m-2,对于高温、酸、碱及氧化降解条件的选择,指导原则中并未明确[12],其降解条件选择可参考药物强制降解的综述文献中推荐的条件进行不断摸索研究。美国FDA审评员,辉瑞、葛兰素史克、阿斯利康等国际制药公司的研发人员认为,5%~ 20% 的降解是可以接受的,对于含量为标示量90% ~110% 的小分子药物,约10% 的降解为最佳。对于酸、碱、氧化降解条件,不同文献推荐的降解条件不同,综合相关报道[2-7]及经验,可推荐的酸碱降解条件:药物质量浓度 0.1 ~ 1.0 mg·mL-1,0.1~ 1mol·L-1盐酸溶液、硫酸或氢氧化钠溶液、氢氧化钾溶液、氢氧化锂溶液等,温度为室温至70℃,降解时间不过3周;对于氧化降 解:药 物 质 量 浓 度 为0.1~ 20mg · mL-1,0.02%~3% 过氧化氢溶液,温度为室温至30℃ ,降解时间不过1周,当反应温度超过50℃ ,过氧化氢的O- O键会断裂形成自由基,加剧不可预测的降解途径的形成,另外,过氧化氢的浓度过高可能导致伪降解产物的生成,使得降解研究的意义变得有限。当反应条件提高到上述降解条件的极限时,药物仍然较稳定或无明显降解产物生成时,不推荐采用更加剧烈的条件继续进行试验[13-14]。
对于固态高温降解,有文献[15-16] 认为与低温时相比,降解温度过高时,化合物的降解途径可能会发生较大的变化,研究此高温下的降解产物将变的意义不大,因此,高温降解的温度通常不宜过高。对于高温降解的终点判定,一方面需要使得药物获得充分地降解;另一方面,美国药品研究与制造企业协会( Pharaceutical Research and Manufacturers of Ameri.ca,PhRMA))认为,高温降解的能量应不低于样品在加速条件下(如40℃ /75%RH,6个月)所接受的能量[17],二者之中的任何一种情况首先达到,即可终止降解。一般可根据Arrhenius方程理解和判断降解条件的选择,即通常情况下,药物在固定湿度下的高温降解动力学过程遵守下式:k=Aexp(-Ea/ RT),其中,k为速率常数,A为频率因子,R 为摩尔气体常数,T为热力学温度,Ea为表观活化能,当化合物的活化能Ea已知时,可以根据此公式推算出不同温度下的降解速率,如表1示。

Ea实际测定需要进行一系列复杂的工作。可采用保守和激进的估算值对试验进行预估,例如Ea采用较激进的19.8 kcal·mol-1时,当某原料药或制剂的加 速 试 验 条 件 为40℃/75%RH放置6个月(180d)时,相当于在70 ℃ 下放置11d(180d ×5.0÷80.6=11d),60 ℃ 下放置27d;当Ea采用较为保守的17kcal·mol-1时,由上表可知,加速条件40℃ /75,相当于70℃下放置17d(180d ×4.0 ÷ 43.2=17d),60℃ 下放置35d;当采用更加保守的Ea(12kcal·mol-1 )时,相当于70 ℃下放置33d(180d × 2.6 ÷ 14.3=33d),60℃下放置56d, 已 有 研 究 报 道 大 多 数 化 合 物 的Ea在20kcal·mol-1 以上[18],所以可采用较为激进的Ea进行预估。需要注意,以上不同温度之间降解速率的换算需要基于降解反应的湿度保持不变,因此,为了方便估算,建议高温降解反应的湿度保持与加速条件一致(如75% RH)。如果没有证据表明更加剧烈的温度会影响药物的降解途径,可以采用更高的温度进行试验,以节省反应的时间,以 Ea 采用18kcal·mol-1 为例,110℃下反应16h 相当于长期条件25℃/60%RH下放置18个月[19]。
强制降解试验中应注意考察降解前后的物料平衡、主峰的峰纯度及主峰和相邻杂质的分离度,制剂的强制降解试验可考虑平行设计空白辅料和原料药降解试验,排除二者对制剂的降解试验干扰。
2、强制降解产物研究
ICHQ1A(R2)指导原则认为,对于创新药和新制剂,虽然强制降解产物的考察有助于确定降解途径、帮助开发和验证适当的分析检测方法,但如果某强制降解产物被证明未在加速或长期稳定性试验中产生,则对该强制降解产物的考察可能是没有必要的。但创新药强制降解下主要降解产物归属的研究,对了解其降解途径,提前发现潜在致突变降解杂质,评估其安全性和毒性等均具有重要意义。 作为主体责任人,国外多数企业均选择对创新药强制降解中的主要产物进行结构确证,以了解药物的降解途径和规律[20]。
2.1 主要降解产物的判定
化学仿制药的降解途径和主要降解产物已有报道。 如抗流感药物帕拉米韦注射液原研药品的主要降解途径/ 降解杂质在日本PMDA审评文件中已有论述[21],其主要降解杂质为羧基手性消旋杂质(C-1异构体杂质);在注射液Q1Q2一致的基础上,其降解途径应与原研药品基本一致,故一般无需再通过对主要降解产物进行归属来重复这些背景知识[22]。创新药的结构新颖,降解途径未可知,对其强制降解条件下主要降解产物进行归属,对了解其降解路线具有重要意义。 目前各国药品监管部门均无对主要降解产物的判定进行论述,主要降解产物的判断也应当结合药物的正式稳定性的研究结果进行判断。 国外业界所报道的判定方法或观点如下[19,23-24]:第一种方法是一条经验性的判断方法(表2) ,具体规定为对固体样品,主要降解产物应至少为0.25% ;对于液体样品,主要降解产物应至少为1% ,总降解产物不超过20% 。 当满足该前提条 件 时, 可根据下文对主要降解产物 进行判定。

该判定方法具有阶段性,规定了不同申报阶段的判定标准。 例如,某原料药在申报临床试验申请( investigational new drug, IND) 阶段,在光照条件下降解 了4.46% , 分 别 为 杂 质A(0.8% ) 、 杂 质B(3.0% ) 、杂质C(0.02% ) 、杂质 D(0.04% ) 、杂质E(0.1% ) 和 杂 质 F( 0.5% ) 。杂 质 C、 D、E的 含量 <0.25% ,直接不作为主要降解产物;杂质 A、F分别占 总 降 解 产 物 的17.9% (0.8% ÷ 4.46% ) 、11.2% (0.5% ÷ 4.46% ) ,大于表中的上限10% ;另外,2 个杂质的含量分别为最大降解产物的26.7%(0.8% ÷ 3. 0% ,大于限度25% )和 16.7% (0.5% ÷3.0% ,小于限度25% ),所以杂质f不应视为主要降解产物,故杂质A、B为主要降解产物,应进行结构确证;但当该药物进行NDA申报时,杂质F应进行鉴定(16.7% ,大于限度10% )。另一种方法将ICHQ3A、3B中的鉴定限与强制降解的鉴定限进行了关联,一般仅针对高温固体降解试验。该判定方法的大致原理:首先,假定高温固体降解试验与加速条件40℃ /75% RH 放置6个月达到了动力学平衡;其次,保守估计若药物在货架期内降解了 2% ,如在强制降解试验中应降解5% ~10% ,经计算,后者是前者的2.5~5倍,取其中较大的倍数5。当以ICHQ3B中的鉴定限为基数再乘以相应的 5倍时,则可以推导出强制降解中的鉴定限(表 3)。
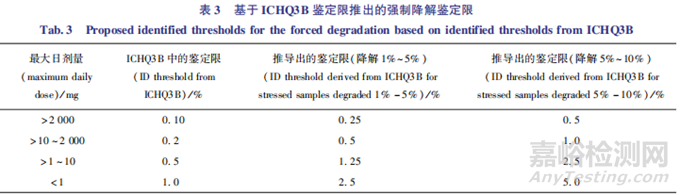
需要说明的是,如表中所示,如强制降解试验仅降解了1%~5% ,是货架期降解的0.5~2.8倍,取其中较大的倍数2.5倍, 则强 制降 解 的 鉴 定 限 为ICHQ3B鉴定限乘以2.5。例如,某片剂最大日剂量800mg,在高温降解 (70℃ ,11 d) 下与加速条件40℃ /75%RH 放置6个月达到了动力学平衡,降解产物量为2%,则该强制降解研究中,主要降解产物为 >0.5% 的降解物。
2.2 降解产物的结构确证
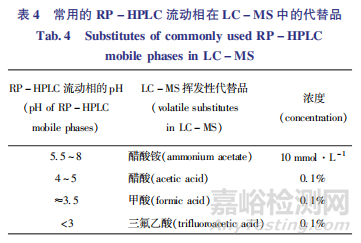
应随着创新药研发的进展不断完善主要降解产物的结构确证研究。反相高效液相色谱(RP-HPLC)是强制降解研究常用的分离手段,经过评估确定的主要降解产物通常采用液相色谱 - 质谱联用仪(LC-MS) 进行结构确证,如主要降解产物降解量足够大,也可通过制备色谱柱对其进行分离,再结合其他技术手段(如核磁共振 NMR、红外光谱IR、质谱MS等)进行进一步确证。由于常用的RP-HPLC液相条件通常含有非挥发性的缓冲盐,故采用LC-MS进行结构确证时,可使用具有相似 pH 缓冲范围的挥发性缓冲盐对其进行代替,如PH 5.5~8的RP-HPLC流动相可在LC-MS采用PH 约为6.8 的10 mmoI·L-1醋酸铵溶液代替,并可以通过添加适当醋酸或氨水调节PH,常用的RP-HPLC的替代流动相见表4。需要注意LC-MS条件下 的 液 相 色 谱 保 留 组 分 与RP-HPLC的对应关系,一般来说,若LC-MS方法中流动相的PH与原始方法能基本保持一致,则二者的流出特征通常非常类似[25]。
2.3 致突变降解产物研究
ICH M 7(R2)主要适用于 创 新 药, 指 出 潜 在 降 解 产 物 包 括 加 速 试 验 和ICHQ1B光照稳定性试验下形成的超过ICHQ3A、3B鉴定限的产物,不论实际降解产物还是潜在降解产物,只要鉴定出了结构,均应对其进行致突变性评估[26],化学降解原理、强制降解研究可帮助选择潜在降解产物。可见,对于创新药而言,对强制降解条件下主要降解产物进行归属有助于识别其可能产生的致突变降解产物,并拟定合理的策略予以预防。例如,芳基酰胺结构类药物(如对乙酰氨基酚) 、含有吡啶基团的药物(如质子泵抑制剂) 、酚类药物(如丙泊酚)分别容易在水解、氧化条件下降解产生致突变警示结构的芳香胺,N-氧化物和醌类药物等,在产品开发过程应注意选择合适的PH,关注产品的包装形式是否需要隔氧、遮光等,应结合长期稳定性试验结果对包括光照强制降解条件下的降解产物的致突变性进行评估,当降解产物最终鉴定为致突变杂质而在实际储存条件下未检出时,可以不进行进一步评估,但需要注意分析方法灵敏度是否能达到要求;当实际储存条件下有检出且高于安全限度时,应该采取控制措施降低风险。
3、小结
药物强制降解研究包括酸、碱、氧化、高温和光照降解5个方面,化学创新药的强制降解研究有利于加强研发人员对其降解途径、机制的理解,并验证所用分析方法的指示能力。对于光照试验条件,ICH指导原则已经有了较明确的规定;对于高温、酸、碱、氧化反应试验,极限条件的确定需要结合药物的结构性质以及一般允许的降解程度去判断,主要降解产物的判断也应当结合药物正式稳定性的研究结果和国际经验,合理的强制降解产物的结构确证加强了对药物降解路线的确定,同时,随着ICHM7(R2)的实施,强制降解研究也对致突变杂质的评估起着重要的作用。
参考文献
[1] ICH, Sability Testing of New Dmg Substanoes ad Produets01A(R2),Internaional Council far Harmonisation[S].2003:1L 2
[2]郭涤亮,浅谈化学药物强制降解试验的设计与开展[J].中国新药杂志,2019,28(20):2468GUO DL,Casiderations on the csign ad implementtion ochemical dmgs sinss testing[」]. Chin New Drug J,2019,28(20):2468
[3] 马骏威,刘涓,刘永辉,等,强制降解试验在药物研发中的应用[J],中国现代应用药学,2020,37(14):1778
MA JW, LlU j, LlU YH, e al. Applieation af foresd degradationtesting in drug developnnt[j]. Chin J Mod Appl Phar , 2020,37(14):1778
[4] BLFSSY M,PATELRD, PRAJAPA'T PN,et af. Devkopment oforol degndaion and sability inieating studies af druss-a nview[j].JPhan Anal,2014,4:159
[5]SINGH S, JUNWAL. M, MODHE, et al, Foredl degrdation stu!ies to assess the stabilily of drugs and products[ 」]. Trae -trendAnal chem, 2013,49:71
[6]SINCH S, BAKSH!M, Guichnoe on conduet of stress tests to determine inherent stability of dmgs [J]. Phamn Technol, 2000,24:1
[7] NGWA G. Foreed degradation sudies as an integml pat of HPlCstability indieating me thod developnent [ j]. Drg Deliv Technal,2010,10(5): 56
[8] PMDA. Levllaxwcin 500 mg [EB0L].[2024 -12-23 ]. hups y//www,info.puln.so.ip'gwintenview/1/80002_6241402G10831004 1F
[ 9] 于佳,杨利红,胡吕勤,等,头孢曲松钠降解试验及降解产物的结构鉴定[J],中南药学,2014,12(2):106
YU J, YANG LH, HU C0,et al, Degnlaian test of cefriawoneselium and simdure iekntifeation af its cegralation proluetsCent Sauth Phamn, 2014,12(2):106
[10]巫世卫,蒋伟哲,陈勇,等,左旋多巴的强降解实验研究[J]中国药业,2008,17(21):20WUI SW, JANG, WZ,CHEN Y,e al. Experinxntnl sukby on freedkgrxlation of levabp]].chin Phamn,2008,17(21):20
WU SW, JANG, WZ, CHEN Y,d al, Experientnl sudy on fneeddegrxlnion of lewabp[I]. Chin Pha, 2008, 17(21):20
[11] FDA. Guidaee far lndudry, INDs for Phase l and ll ShudiesChemidry,Maufaeturing,and Cntrls infermatin, Food andDmg Administmtion [EB/0L].[2024-12 -23 ]. hip://www, fda. gav/dewnlaads/Dmgs/GuidaneeComplimneeRegulatoryInomnation/Guidanee//uem070567. pdf
[12] ICH, Subility Testing: Photestability Tesing af New Drug Substanes and Pruueks 0lB, itemational Couneil for Hamnisati.an[S]. 1996
[13]MAHESWAR AN R, FDA pespedives: scientife oasidematians offoreed degmdntion studies in ANDA submissions 【J]. PhamnTechnal, 2012,36(5): 73
[14]KLICK S, MUISELAAR PG, WATERVAL I. Towand a generieapproach for siress testing of drug sbstaees ad dng produets[1].Pharm Tehnol, 2005,29(2): 48
[15] DORMAN DE,LORENZ L,0CCOLOWTZ JL.. bsalatian andstruture elucidation of he majar degradatien produets of eefnelarin the salid stnte[」]. J Phann Sei, 1997,86:540
[16] OLSEN BA, PERRY FM, SNOREK SV, Aeoelemted canditionsfor siabilily asessnxnt af bulk ad fammlated ceaekr mna.hydmte[j]. Pharm Dev Tehnol, 1997,2: 303
[17] REYNOLDS DW, FACCHINE KL, MULLANEY IF. Availble guid.anxs: anel best praciesss far cxuhu: ting forsd cegralntion shudies[]].Phanm Techal, 2002,26(2): 48
[18]BAERISCHI SW, ALSANTE KM, REED RA. Dngs anl the Pharnmoesicl Scieees:Phmnoticl Sirss Teding: Pnigian DrugDegmlaicn[M].2011,210:10[19]AISANTE KM, ANDO A, EROWNR. The rle af degralant pr.fling in aetive phamceutical ingnlients ad dng produets[]]Ahv Dng Deliwer Rev,2007.59:29
[20]ALSANTE KM, BAERTSCHI SW, MARTIN L. A stress kestingbenchmaking stuy[J].Pham Technol, 2003 ,27:60
[21]PMDA. RAPIACTAs fr Intraenous Dp lhsion 150 mg; 300 mg[EBVOL].[2024-12-23]. htps://www. infa pmxl. sr. jpygwinkerview/1/340018_6250106A1032 1_011 1F
[22]黄晓龙,浅谈强制降解试验[EB/OL].[2020-02-23].hitlp://www.oe.org.en/dzkw, do? methad = largePage& id =60888c33 23.5ee4l
HUANG XL. Discusion on Foreed Degradation [ EB/0L].[2020-02-23].hilp://www. ede. ag. cn/dzkw. do?method =largePage&id=60888c3323e5e41
[23]KIFNMAN MH, ELDER D, TEASDALE A. Simtegies io aldnssmutngenie imywurities derived fom dgrad,tion in dnug sulstanees andnug prahuts[J]. 0rg Press Res Dev, 2015, 19: 1447
[24 DOW LK, HANSEN MM, PACK BW, The sssessnent af impuri-ties for genotekie potentinl and subsequent cantrol in dmg sub.stanoe and drug proluet[I]. J Phann Sei, 2013, 102: 1404
[25]李敏,药物降解的有机化学[M].北京:化学工业出版社,2019 : 207
[26]IM. Oganie Chemistry of Dng Degralaian [ M]. Beijing:Chemical lndustry Press,2019:207ICH, Assessnent and Contral of DNA Reactiw ( Mutagenie )Impurities in Pharmaceutieals to Limit Potential Carcinogenierisk M7(R2),Itematianal Couneil for Hanonisation[S].2023

来源:药物分析杂志


