您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2015-07-01 23:02
一、医疗器械适用的RoHS2.0要求
我们常说的RoHS即欧盟《限制某些有害物质在电子电器设备中的使用指令》,现在该指令已是第2版,所以业界一般将其简称为RoHS2.0。
RoHS适用的产品对象是所有进入欧盟市场的“电子电器设备”。在新版的RoHS2.0中,医疗器械属于新纳入的产品类别,需要符合RoHS2.0要求。
需要明确的是,并非所有医疗器械都需要满足RoHS要求,只有那些属于RoHS2.0指令定义的“电子电器设备”范围的医疗器械,才需要满足RoHS要求。
RoHS2.0对“电子电器设备”的定义为:指依赖电流或电磁场才能正常工作的设备,以及用于产生、转换和测量上述电流或电磁场的设备,且设计使用电压交流不超过1000伏、直流不超过1500伏。
所以,只要符合上述定义的医疗器械,都属于需要符合RoHS要求的“电子电器设备”。请注意,RoHS2.0将“植入式医疗器械”排除在外,即使它们符合上述“电子电器设备”的定义,这类医疗器械也无需满足RoHS2.0的要求。
通常,业界将耗电的医疗器械简称为“有源医疗器械”。虽然这个不很准确,为行文方便,本文还是采用这个术语。
2. RoHS2.0对有源医疗器械的生效时间
RoHS指令对不同类别的电子电器设备规定了不同的生效时间。
对于医疗器械的生效时间,RoHS2.0进一步分别针对一般的医疗器械(符合欧盟93/42/EEC指令第1(2)款第a点的定义的)和“体外诊断医疗器械”(符合欧盟98/79/EC指令第1(2)款第b点的定义的)进行了规定。
对于一般医疗器械,RoHS指令生效的时间是2014年7月22日,即从2014年7月22日开始投放欧盟市场的一般医疗器械必须符合RoHS2.0的要求了。
对于“体外诊断医疗器械”,其生效时间则为2016年7月22日。
所以,现在我们必须关注的是普通有源医疗器械的RoHS符合性。
3. RoHS2.0对有源医疗器械的适用要求
RoHS2.0对有源医疗器械的适用要求罗列如下:
产品中含有的“铅”、“镉”、“汞”、“六价铬”、“多溴联苯”、“多溴联苯醚”等六类物质不能超过限值。对这六类物质的限值除了“镉”为“0.01%”外,其余五类的都是“0.1%”。而且,对这些有害物质的限值都是从“均质材料”层次进行的,极为严格。这个要求是RoHS2.0指令的基本要求。
此外,RoHS2.0对制造商还有如下要求:
制造商要按照第768/2008/EC决定的附件II的模式A制订技术文档,实施内部生产控制程序,或确保其得到实施;
制造商应制订欧盟合格声明,并在成品上加贴CE标志;
制造商应留存技术文档和欧盟合格声明到电子电气设备投放市场后10年;
为保持符合性,制造商应确保系列生产执行该程序。应充分考虑产品设计变更、特性变更,以及对电子电气设备合格声明引用过的协调标准或技术规范变更;
制造商应登记不符合的电子电气设备和产品召回,并通知相关经销商;
制造商应确保自己的电子电气设备带有型号、批次和序列号或其他有助于识别产品的要素;在产品的形状或性质不允许时,在包装或产品随附文件中提供;
制造商应将公司名称、注册的商标名称或注册商标标识及联系地址在电子电气设备上标明,不可行时,在产品包装或随附文件中提供。地址一定要指明能够联系到制造商的一个点;在欧盟其他适用法规中包含至少同等严格程度的加贴制造商名称和地址的条款时,适用那些条款;
当制造商认为或有理由相信投放市场的产品不符合本指令时,应立即采取必要的纠正措施使该电子电气设备符合、适宜时撤回或召回;而且,应立即将相关情况通知产品所投放成员国的主管部门,特别应告知不符合情况、应采取纠正措施的详细信息;
应主管部门的合理要求,制造商应以主管部门能理解的语言向其提供可用于证明电子电气设备符合本指令的所有信息和文件。应主管部门要求,他们应与主管部门合作,针对其投放市场的产品采取确保符合本指令条款的任何措施。
二、医疗器械企业CE认证与RoHS2.0符合性的关系
欧盟医疗器械指令93/42/EEC之条款4(5)明确规定:如果医疗器械也需要符合其他关于别的因素的指令,且也要求加贴CE标志时,(产品上)加贴的CE标志应表明该医疗器械也满足这个指令的条款要求。
欧盟体外诊断医疗器械指令 98/79/EC之条款4(5)明确规定:如果医疗器械也需要符合其他关于别的因素的指令,且也要求加贴CE标志时,(产品上)加贴的CE标志应表明该医疗器械也满足这个指令的条款要求。
也即是说,有源医疗器械上加贴了CE标志,表明制造商在声明该医疗器械产品既符合对应的医疗器械指令的要求,也符合其他适用的CE标志指令的要求,如RoHS2.0的要求。
表述的更容易理解一点就是:
不符合RoHS指令的有源医疗器械产品,不能加贴CE标志;
加贴了CE标志但不符合RoHS的有源医疗器械产品,进入欧盟的话,是违法的,存在极大的风险;
不符合RoHS指令的有源医疗器械产品,不应该通过医疗器械CE认证。也即意味着对医疗器械的CE认证,应该审核RoHS符合性,并确定产品符合RoHS要求才允许通过认证。
对于医疗器械认证机构而言,让不符合RoHS2.0要求的有源医疗器械产品通过CE认证将具有极大风险。如果经其认证的有源医疗器械在欧盟市场上被监管部门抽查,且结果是不符合RoHS2.0的要求,那么,认证机构的资质可能因此而受影响,因为产品的CE标志上带有认证机构的编号。
因此,欧盟有源医疗器械的认证机构在开展CE认证时,应该审核产品的RoHS2.0符合性。
三、医疗器械CE认证审核员如何把握产品对RoHS2.0的符合性
准确把握与CE标志相关的RoHS2.0要求
a)产品中有害物质不能超标
b)产品需制订并随附技术文档,技术文档需符合EN 50581标准的要求
c)产品随附符合性声明,声明需符合RoHS2.0附件VI的格式和内容要求
d)产品加贴CE标志,标志格式需符合欧盟对CE标志的要求
2. 重点关注产品有害物质超标的主要风险
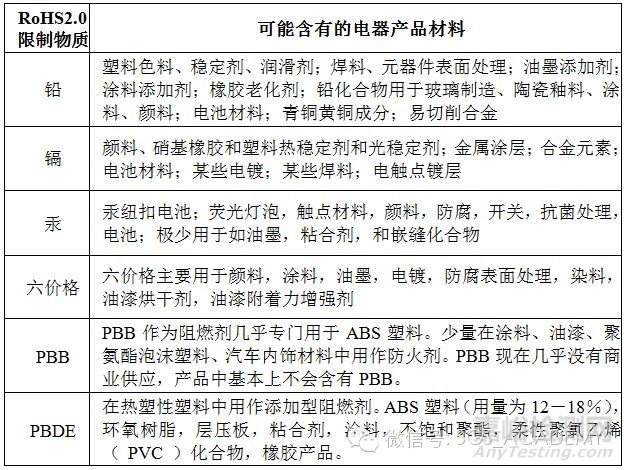
a) 风险材料:是导致产品有害物质超标的最大风险。但有害物质只会存在于某些材料中,见下表:
b)风险工艺:某些工艺可能导致有害物质产生、转移/污染或浓度的变化
c)人为失误:如混料/混货:回用塑料混料;非常多,风险最大,同时做两种产品的要重点关注
3. 把握关键风险过程
如上所述,有害物质超标的风险,最主要的来源是原材料中有害物质超标。所以,一下过程的控制效果是关键,审核员应重点关注这些过程:
物料确认过程:必须确认公司拟采购的物料是经过充分验证符合RoHS的。
采购过程:公司采购的产品只能是经过确认的产品,供应商也必须是经过确认的。
来料检验:公司应该具备一定的有害物质检测能力,如配备XRF类快速测定仪。
仓库管理:确保物料的正确标识、正确发放,确保不发生混料。
生产过程:确保物料的正确领用,确保不发生混料。
产品检测:一般客户要求产品进行年度第三方RoHS检测。
技术文档:确保公司已按照EN 50581要求制定了技术文档。
符合性声明:确保公司已按照RoHS2.0附件VI的格式和内容制定了符合性声明。
CE标志:确保CE标志格式和加贴的正确性。
来源:SGS


