您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-03-10 10:16
注射剂的无菌保证工艺
无菌制剂包括直接注入体内的注射剂或直接用于创面及黏膜的非胃肠给药制剂。 由于这类制剂的特殊给药部位,无菌制剂的质量及安全风险显著高于其他类别制剂,必须保证最终产品的无菌性。
一、注射剂的无菌保证工艺分类
注射剂无菌保证工艺是指为实现规定的无菌保证水平所采取的经过充分验证后的灭菌(无菌)生产工艺。目前,注射剂的无菌保证工艺主要有两种
1、终端灭菌工艺(terminal sterilization process) 在控制微生物污染量的基础上,在药品灌封后,通过湿热灭菌方式除菌。 一般来说,本方法成本低,无菌保证水平高,适宜于大容量注射剂的灭菌。
2、无菌生产工艺(aseptic processing) 是指以防止污染为目的,在无菌系统环境下,通过除菌过滤法或无菌操作法,消除导致污染的各种可能性来保证无菌水平。 无菌生产工艺和终端灭菌工艺具有不同的系统要求、不同的除菌方法和不同的无菌保证结果,这是由于无菌生产工艺对环境系统的要求高,且影响无菌操作的因素多而使得无菌保证水平比终端灭菌工艺低。
无菌生产工艺一般适宜于粉针剂,亦可适宜于临床需要,但不能进行终端灭菌的小容量注射剂。 目前评价无菌生产工艺是否有效,多注重无菌生产工艺的设计是否合理,所用的设备与工艺是否经过充分的验证,在此基础上,切实按照验证后的工艺进行生产,以保证灭菌(无菌)工艺的可靠性。
“无菌保证水平”(sterility assurance level,SAL)为产品经灭菌( 除菌)后微生物残存的概率。SAL是评价灭菌(无菌)工艺的效果的重要指标。该值越小,表明产品中微生物存在的概率越小。 为了保证注射剂的无菌安全性,国际上一致规定,采用湿热灭菌法的SAL,不得大于10的(-6次方),即灭菌后微生物存活的概率不得大于百万分之一;而采用无菌生产工艺的产品,其SAL一般只能达到10的(-3次方),可见非终端灭菌制剂存在微生物的概率远远高于终端灭菌制剂,故仅限于临床必须注射给药而确实无法耐受终端灭菌的产品。
二、注射剂灭菌工艺的选择
注射剂的灭菌是保证制剂质量和用药安全性的重要工艺步骤。 为保证灭菌的有效性和制剂的无菌保证水平,注射剂灭菌工艺的选择原则是:优先选择无菌保证水平高的终端灭菌工艺,只有在经充分的工艺研究证明无法耐受终端灭菌工艺的前提下,才选择非终端灭菌工艺。
为此,欧盟“欧洲药品评价局(EMEA)”专门制定了规范性文件“灭菌方法选择决策树"(decision trees for the selection of sterilization methods,CPMP/QWP/054/98),分别规定了水溶液型无菌制剂和其他无菌制剂灭菌方法选择的原则。见下图。
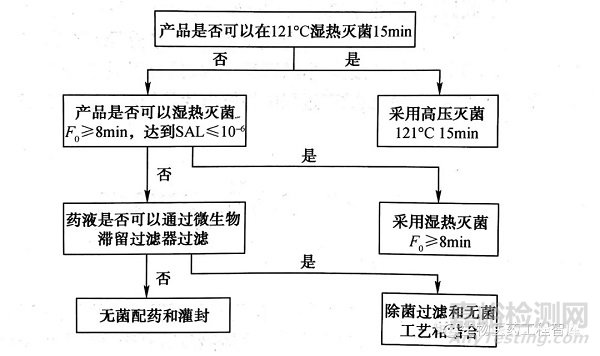
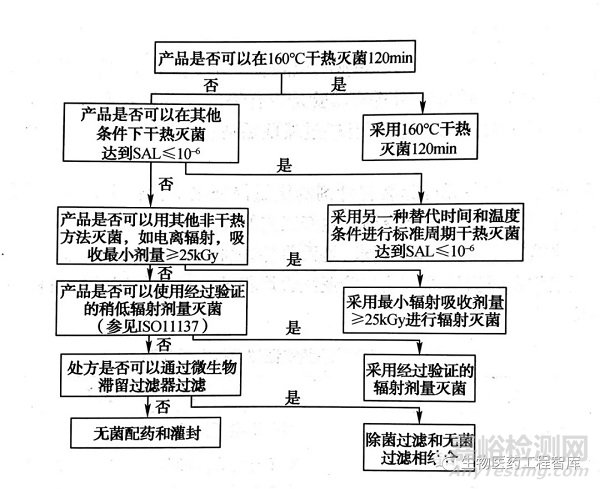
注射剂生产过程中,除应选择恰当的灭菌工艺外,还应对灭菌前产品中污染的微生物严加监控,并采用各种措施降低微生物污染水平,确保终产品达到无菌保证要求。此外为判断灭菌工艺对产品质量的影响,应进行灭菌前后的质量对比研究,且考察项目需全面,相关方法需验证。
应该指出的是,除了取决于活性成分的性质外,药品能否耐受终端灭菌工艺,很大程度上还与所用的溶剂、辅料、氧气、原料药中的杂质等密切相关。灭菌前各生产步骤的工艺研究和控制,对产品能否采用终端灭菌工艺具有重要的影响。
如国内外的生产研究和实践证明,复方氨基酸注射液中半胱氨酸在121℃会分解,如果将溶液中的溶解氧浓度降低到0.1ppm,含有胱氨酸等18种氨基酸的复方氨基酸注射液至少可以在121℃灭菌10分钟以上(F0值>10)。 但如果不控制溶解氧,即使加人500mg/L的亚硫酸盐类抗氧剂,也只能在110℃灭菌30分钟以上(F,值<3)。
三、无菌保证工艺的技术要求
1、大容量注射剂
(1)应采取终端灭菌工艺,建议首选过度杀灭法(F0≥12),如产品不能耐受过度杀灭的条件,可考虑采用残存概率法(8≤F0<12),但均应保证产品灭菌后的SAL不大于10的(-6次方)。原则上不应采用其他F0值小于8的终端灭菌工艺。
(2)如产品不能耐受终端灭菌工艺条件,应尽量优化处方工艺,以改善制剂的耐热性。如确实无法耐受,则应考虑选择其他剂型,而非大容量注射剂。
(3)应进行规范的灭菌工艺验证,部分验证工作可结合生产线验证一并进行。
2、粉针剂
采用无菌生产工艺的粉针剂,应能保证SAL不大于10(-3次方),这主要依赖于无菌生产工艺是否严格按照药品生产质量管理规范(GMP)的要求进行生产与验证。
(1)冻干粉针剂:冻干粉针剂无菌生产工艺验证中的设备验证、环境监测是其生产线GMP要求的常规内容;培养基灌装验证是对设备、环境以及人员操作的一种系统验证;是判断无菌保证水平的关键手段。
常规的工艺验证试验包括:
①培养基模拟灌装验证试验最少在线灌装3批,每瓶产品均应进行无菌检查,每批的批量和判断该试验是否合格的标准见下表。
②除菌过滤系统适应性验证试验包括过滤系统相容性测试、过滤前后滤膜完整性测试、滤膜的微生物截留量QOS商灭测试。
(2)无菌分装粉针剂:无菌分装粉针剂的质量保证主要依赖于无菌生产线的基本条件和对生产工艺各环节严格的质量控制。生产工艺的控制和验证要求对不同的无菌分装产品是一致的。 严格执行GMP 的有关要求,是无菌粉针剂生产的重要质量保证。
工艺验证工作主要为培养基灌装验证试验培养基模拟灌装试验的目标是零污染,灌装的批量与评价标准见下表。

3、小容量注射剂
(1)应首选终端灭菌工艺,相关技术要求同大容量注射剂。
(2)如有充分的依据证明不能采用终端灭菌工艺的品种,且为临床必须注射给药的品种,可考虑采用无菌生产工艺,相关技术要求同冻干粉针剂。
(3)对于采用无菌生产工艺生产的小容量注射剂,生产线的验证应结合无菌生产工艺进行。
在剂型选择的研究中,为判断灭菌工艺对产品质量的影响,应进行灭菌前后产品质量对比的研究,且应注意考察条件和方法的合理性,考察项目需全面,相关分析方法需验证,同时研究用样品应具有代表性。
容器的密封性对于无菌产品在有效期内保持无菌性能具有重要作用,在工艺研究、包装材料的选择以及稳定性研究中,应加强对容器密封性的考察。
灭菌(无菌)生产工艺验证
灭菌工艺的验证并提供完整系统的工艺验证资料是保证注射剂灭菌的可靠性的重要环节。 工艺的验证包括对生产环境、设备条件是否符合设计要求的验证,以及对采用的灭菌工艺是否可确保制剂的无菌保证水平的验证。 对于直接接触药品的包装材料及容器生产设备等的灭菌亦应进行验证。
一、 终端灭菌无菌药品的灭菌工艺验证
生产设备已经完成安装和运行确认是灭菌工艺验证的前提。 验证试验主要包括四项内容:空载热分布、满载热分布、热穿透试验和微生物挑战试验,前三项试验主要是对灭菌设备进行验证。 终端灭菌工艺验证实际上是通过上述四部分试验过程来确认生产的药品可以达到规定的无菌保证水平。
1、空载热分布试验 该试验主要是确定灭菌器腔室内温度分布的均匀性和重现性,应至少连续运行同一个完整的灭菌程序3次。由于很多情况下一台灭菌设备可能运行多种灭菌程序,可以根据灭菌设备的构造特点,在保证科学性的前提下,选择一个有代表性的灭菌程序进行空载热分布试验。
2、满载热分布试验 该试验主要是确定在满载的情况下灭菌器腔室内温度分布的均匀性,个别位置的温度与平均温度的差异,是否存在冷点和热点。应根据空载热分布试验的结果确定试验方案。 在规定的灭菌程序运行达到灭菌温度时,个别点的温度与平均温度有显著差异时,该点为冷点或热点。满载热分布试验发现的冷点或热点为下阶段进行热穿透试验重点考察的位置。应至少连续运行3次完整的灭菌程序,考察灭菌设备的均匀性和重现性。
3、热穿透试验 热穿透试验主要是在灭菌过程中获取不同位置的产品实际达到的温度和F0值,从而了解不同位置之间,以及与日常运行时灭菌设备记录的温度与F0值之间的差异。
应根据满载热分布试验的结果制定试验方案,至少进行最大和最小装载条件下的热穿透试验。 若满载热分布试验中发现有冷点或热点,应重点采集冷点或热点位置的热穿透数据。一个灭菌工艺的试验应至少运行3次灭菌程序以证明其重现性。
热穿透试验方案应详细说明所用的温度传感器的数量及其安装分布方式,装载的形式,实验采用的是真实产品还是模拟产品,各项试验运行灭菌程序的次数。如果采用模拟产品,应有数据证明模拟产品与真实产品没有热穿透差异。
为了预判可能不易达到均匀受热的部位,有目的地安装温度传感器并确定试验的次数,应根据灭菌设备的特点进行分析。 用于验证的温度传感器和温度数据采集记录系统应符合要求。 仪器测量温度的误差至少不大于灭菌温度参数允许波动范围的 1/3。 如灭菌温度范围为121℃土1.5℃,则温度采集系统的误差不能超过0.5℃。
国际上并没有明确地规定各项试验所需要的探头数量,但原则是根据设备的设计和工作原理合理地判断可能的不易达到均匀受热的部位,有重点地安装探头。 温度测试所使用的温度探头在验证前后都应经过标准温度计校验,标准温度探头每年都应送法定计量单位校验。
4、微生物挑战试验
该试验主要是最终确认灭菌工艺对挑战用生物指示剂的杀灭效果,通过试验证明在确定的灭菌参数允许的最低灭菌条件下,应用该灭菌工艺能将符合灭菌前污染微生物数量和耐热性限度的产品中的微生物杀灭至存活概率不超过百万分之一。
(1)生物指示剂:生物指示剂是一类特殊的活微生物制品,用于灭菌验证的生物指示剂一般是细菌的孢子,可通过采购或自行制备得到。
(2)试验方案:应根据热穿透试验的结果设计试验方案。 若热穿透试验证明不同位置的产品间、不同装载量间、不同装量规格间的热穿透特性有显著差异,应至少选择灭菌F0值最低的位置、装载量和装量规格( 即最差条件)进行微生物挑战试验。
微生物挑战试验的形式通常是将生物指示剂定量地加入到产品中,制备成带有确定数量微生物孢子的受试产品。再将产品安放在灭菌设备的特定位置,以尽可能低的灭菌参数运行灭菌程序后,对受试产品进行无菌调查,通过计算预定限度的微生物污染产品经灭菌工之处理后微生物的残存概率,对灭菌工艺的灭菌效果进行验证。
若经过灭菌后所有受试产品的无菌检杏均为阴性,则证明该灭菌工艺能将火菌前每瓶污染有微生物N0。且耐热性不招过D0的微生物杀灭,可保证产品中微生物的存活概率不超放百万分之一。
另外,对受试产品进行无菌检杳的方法应通过验证;测定生物指示剂在产品中D值的方法应通过验证。对于最低灭菌F0值不小于12分钟(即过度杀灭灭菌工艺),且已通过热穿透试验证明具最低F0值的灭菌工艺,可不进行微生物挑战试验。对于最低灭菌F0值小于12分钟的灭菌工艺(即残存概率灭菌工艺),应进行微生物挑战试验。
5、灭菌工艺验证试验结果的评价 根据空载热分布试验结果(在灭菌状态下各温度探头的温度差异),可以判断灭蘭器腔室内温度是否均匀。 根据装载热分布试验(在灭菌状态下室内各处温度与平均温度的差异),判断是否存在冷点与热点。
热穿透试验是验证产品能否达到预定的无菌保证水平的最重要试验之一。通常将F0平均值减3倍标准差F0mean-3std≥12分钟的灭菌工艺定义为过度杀灭灭菌工艺,将F0mean-3d<12分钟的灭菌工艺定义为残存概率灭菌工艺。
对于残存概率灭菌工艺,热穿透试验产品的F0平均值加/减3倍标准差应符合灭菌工艺F0的注册标准。如注册标准F0值为8.5~16.5分钟,则8.5分钟≤Fmean-3stdomean+3std≤16. 5 分钟。 通过验证,应确定热穿透试验F0值的平均值与灭菌设备显示的F0值的允许差异。微生物挑战试验受试样品的无菌检查结果均应为阴性。
二 、非终端灭菌无菌药品的工艺验证
如果药品的热稳定性很差,难以进行最终灭菌,不得不采用无菌工艺进行生产。无菌生产工艺的特点决定了其产品的无菌保证水平远低于终端灭菌产品。 目前国际上较普遍的非最终灭菌无菌产品的无菌保证标准是在95%置信限下产品中微生物残存概率不超过0.1%。
过滤除菌工艺是常用的无菌生产工艺,其工艺验证包括除菌过滤系统验证、培养基模拟灌装验证、过滤前后的滤器完整性测试等。
1、除菌过滤系统验证
(1)滤膜的完整性测试:采用起泡点试验或扩散流量试验进行滤膜过滤前后的完整性
(2)滤膜的相容性测试:包括过滤溶出物的测定、滤液同滤膜是否可能发生相互作用(如滤液的pH、色泽、浊度等是杏发生改变).此项工作一般可由滤器生产企业协助完成。
(3)滤膜对灭菌条件的适应性:考察湿热火菌条件对滤膜的影响,比如有无不溶性微粒出现等。
(4)滤膜的有效使用时间测试:尽管购头的滤膜有有效期的说明,但是由于所过滤药液的性质不同,有效使用时间也会有差别,政米用拟过滤的产品进行考察。
(5)滤膜的微生物截流堇测氓:址仃沫困的过滤挑战试验,因为滤膜的过滤能力可能同药液的性质有关(比如黏度等),故一般采用药液进行。常用生物指示剂为缺陷假单胞菌。
2、培基模拟灌装验证, 无菌生厂工艺应通过培养基模拟灌装验证。验证的基本原理是:采用一定量微生物的培养基溶液模拟整个正常生产过程(即溶液配制、除菌过滤和接收、灌装、密封等过程),验证无菌灌装操作的可靠性。
将模拟生产的产品,按《中国药典》无菌检查的培养条件培养足够时间,并逐瓶检查产品有无染菌。
3、容器密封系统验证 用物理或微生物挑战的方法测试容器密闭系统的密闭性,来保证无菌药品在有效期内的无菌性。具体是采用3个不同批号的容器进行无菌培养基灌装,并成功地通过微生物浸侵试验。
其中要关注在已确定的关键工艺参数范围内进行测试,使用的容器应经过正常生产过程中最大次数的清洗和灭菌(以模拟最恶劣条件),考证新生产的样品以及不同留样条件下的稳定性样品是否能通过微生物浸侵试验。
对于无菌分装工艺,质量保证主要依赖于无菌生产线的基本条件和对生产工艺各环节的严格质量控制。 工艺验证主要进行培养基灌装验证试验。 另外, 工艺验证中还应加强对灭菌前药品的染菌水平、所染菌耐热性等的密切监控,提供相关研究及验证资料。

来源:Internet


