您当前的位置:检测资讯 > 监管召回
嘉峪检测网 2022-02-10 13:44
1.新版MDR和IVDR概述
2021年5月26日,欧盟新版医疗器械法规(EU)2017/745(以下简称MDR法规)和新版体外诊断试剂法规(EU)2017/746(以下简称IVDR法规)正式生效。作为医疗器械和体外诊断试剂的总领性法规,MDR和IVDR对医疗器械和体外诊断试剂(以下统称为医疗器械)的监管机构做出了详细的规定。
2. 欧盟医疗器械监管机构介绍
欧盟由27个成员国组成,设有欧盟议会、欧盟理事会、欧盟委员会等组织机构。欧盟议会的环境、公共卫生和食品安全委员会下设欧洲疾病预防与控制中心、欧洲药品管理局(European Medicines Agency,EMA)等机构,但EMA并不负责医疗器械的监管活动。根据MDR法规和IVDR法规,参与医疗器械注册审评和监管活动的组织机构主要包括:公共卫生部、医疗器械协调小组、医疗器械主管当局、公告机构与各国主管当局等[1,2],见图1。
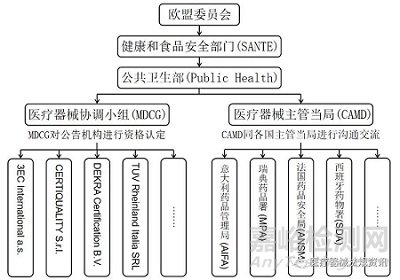
图1 欧盟医疗器械监管机构图
2.1 公共卫生部
欧盟委员会通过起草和执行法规、实施政策、制定欧盟预算并监督执行等行动达成欧盟的整体利益。欧盟委员会的健康和食品安全部门(Health and Food Safety,SANTE)主管欧盟的食品安全和公共卫生,其下设的公共卫生部(Public Health)分管欧盟的制药行业、疾病预防与控制、医疗器械行业和公共卫生系统等。其中,与医疗器械监管相关的组织主要为医疗器械协调小组(Medical Device Coordination Group Working Groups,MDCG)和医疗器械主管当局(Competent Authorities for Medical Devices,CAMD),两者负责管理欧盟各成员国的医疗器械相关机构并开展沟通交流活动,包括各成员国主管当局和利益相关者,如专业组织、患者和医生组织、标准化机构和公告机构等[3]。
2.2 MDCG
公共卫生部(Public Health)下设的医疗器械协调小组(MDCG)由各成员国的医疗器械及体外诊断试剂专家共同组成,主要协助医疗器械相关法规的执行,并负责讨论医疗器械行业的关键问题,如公告机构的监督和标准化、市场监管、国际问题、新技术和临床研究等内容。根据MDR法规和IVDR法规,MDCG的职责具体为受理公告机构的资格认定申请,与公告机构商讨合格评定程序实施相关的问题,进行欧盟医疗器械数据库(EUDAMED)的建立与管理等。
2.3 主管当局
公共卫生部(Public Health)下设的医疗器械主管当局(CAMD)负责促进MDR法规和IVDR法规在各国的实施,就法规执行过程中的问题进行统一答疑,并在适当的时机开展培训交流会议,以帮助各成员国充分理解法规理念并统一贯彻实施。各成员国主管当局与经过资格认定的公告机构一同参与医疗器械的监管,但不负责医疗器械申报资料的审评。以法国主管当局为例,法国药品安全局(ANSM)对医疗器械及体外诊断试剂进行市场方面的监管[4],授权临床试验、对制造商进行现场检查、检查已上市医疗器械的一致性、促进创新等,以确保上市医疗器械的安全性和有效性。
2.4 公告机构
根据MDR法规及IVDR法规,公告机构应当经过欧盟相关部门(即MDCG)的资格认定,结果将公示在NANDO信息系统中。截至2022年2月9日,欧盟已批准6家负责体外诊断试剂申报资料审评的公告机构,27家负责医疗器械申报资料审评的公告机构,30多所公告机构分布于欧盟各个成员国内[5]。MDR法规附录七规定,公告机构负责审评医疗器械/体外诊断试剂申报资料,并对制造商进行质量管理体系审核等。若拟申报医疗器械/体外诊断试剂符合要求,公告机构将出具合格评定证书[1]。制造商取得合格评定证明并准备好符合性声明和CE标志后,产品方可上市销售。
3. 总结
欧盟是一个联邦性组织,其医疗器械监管机构的组成和职责有特殊之处。结合图1,MDCG和CAMD是欧盟下属的医疗器械监管机构,其中MDCG负责公告机构的资质认定,CAMD负责管理与协调各国的医疗器械主管当局,两者皆不负责具体的医疗器械注册审评工作。作为第三方评审机构,公告机构则负责注册申报资料的技术审评工作,而现场检查及产品上市后监管等由各国的医疗器械主管当局负责,两者在具体的医疗器械注册审评流程中各司其职。

图2 欧盟医疗器械和体外诊断试剂的注册流程
如图2,医疗器械制造商在欧盟申请医疗器械上市销售时,首先会将注册申报的技术文件提交给经过资质认定的公告机构,公告机构审评资料,同时制造商/委托商所在地国家的医疗器械主管当局开展现场检查(如需要),若审评合格,则由公告机构出具合格评定证书。其次,收到合格评定证书后,制造商会自拟一份符合性声明并贴上CE标志,声明所涵盖设备已满足法规规定的要求,此时医疗器械产品即可在欧盟上市销售。此外,制造商/委托商所在地国家的医疗器械主管当局还负责临床试验授权,并对已上市产品进行日常的监督管理。
【参考文件】
[1]European Commission. Regulation (EU) 2017/745 on medical devices (MDR)[EB/OL]. (2017-04-05) [2022-02-09].
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745&qid=1634610433796
[2]European Commission. Regulation (EU) 2017/746 on in vitro diagnostic medical devices(IVDR)[EB/OL]. (2017-04-05) [2022-02-09].
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0746&qid=1634610433796
[3]European Commission. Medical Devices - Dialogue between interested parties[EB/OL].(2021-01-28)[2022-02-09].
https://ec.europa.eu/health/md_dialogue/overview_en
[4]ANSM. Who are we[EB/OL].(2021-01-27)[2022-02-09].
https://ansm.sante.fr/qui-sommes-nous/nos-engagements/une-agence-au-service-des-patients
[5]European Commission. Medical Devices - Notified Bodies[EB/OL]. (2021-02-11) [2022-02-09].
https://ec.europa.eu/health/md_topics-interest/notified_bodies_en

来源:Internet


