Question 1:亚太五国平均的注册周期和官费报价?
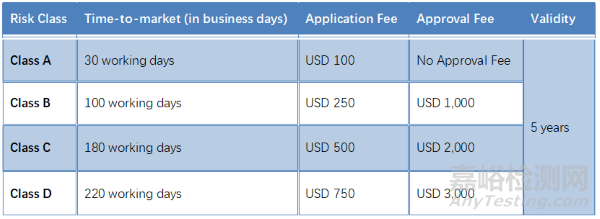
马来西亚
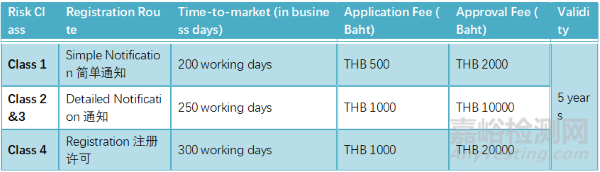
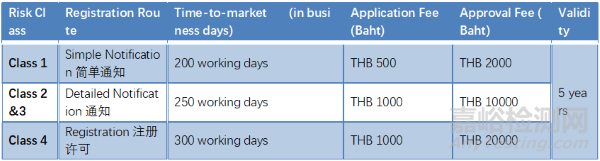
泰国
新加坡
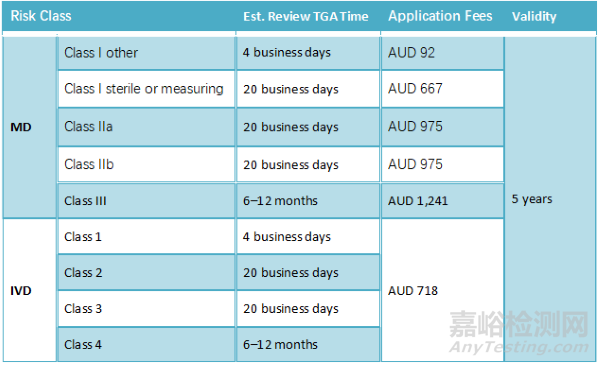
澳大利亚
新西兰:
Official Timeline & Fee
Time to Market
对于WAND名单没有审查程序,只是一个通知(Notification)如果一个制造商是新进入市场的,主办方需要先将该制造商加入WAND数据库,大概5个工作日左右
Application Fee
FREE
在WAND上市不需要缴纳官费
Ouestion 2:亚太五国对于经济运营商、进口商、分销商等有什么要求,需要什么资质?如果产品注册完发生需要更换,需要什么资料或者是否需要重新走注册流程?
马来西亚
● 根据《医疗器械法》第15条,本地机构(如本地制造商、授权代表(ARs)、进口商和分销商)必须从MDA获得许可证。
● 进口商:有执照的进口商必须从当地代表获得LoA,才能将设备进口到马来西亚。进口商向MDA提供此信,作为他们投放市场的产品的通知。
● 经销商:有执照的分销商必须从制造商或当地代表获得LoA,以便在马来西亚分销设备。分销商向MDA提供此信,作为他们在市场上投放产品的通知。
泰国
● 进口商:进口商是指将医疗器械带入泰国或订购医疗器械的实体。进口商必须在泰国FDA注册。
● 分销商:分销商是指为商业目的向他人销售、分配、处置、交换、借出、租赁、租购或转让权利或财产的实体。
新加坡
● 在新加坡制造、进口或以批发方式供应MD/IVD需要许可证。许可证申请指南见GN-02《医疗器械制造商、进口商和批发商许可证指南》。
● 进口商:有执照的进口商是唯一被授权向新加坡进口IVDs的公司。器械的持牌进口商必须由注册人添加到MEDICS的器械注册中,同一产品可以有一个以上的进口商被授权。
● 经销商:在新加坡以批发方式(包括出口)分销MD/IVD需要有批发商的许可证。
澳大利亚
● 进口商:在澳大利亚,每个进口商(即赞助商)都需要单独注册该设备。这包括进口器械用于临床实践的卫生专业人员。
● 分销商:在澳大利亚,没有要求指定一个分销商来注册或销售。一旦产品被列入ARTG(即注册),制造商可以通过分销商或直接向澳大利亚终端用户销售。
新西兰
● 进口商:进口商可以是一个子公司、代理商、"私人标签 "供应商、平行进口商或直接进口商。
Question 3:Q3:亚太五国如何进行医疗器械分类及不同分类下有何不同的监管要求?
马来西亚
● 在马来西亚,医疗器械和IVDs由卫生部下属的医疗器械管理局(MDA)监管。监管框架以《2012年医疗器械法》(737号法案)、《2012年医疗器械管理局法》(738号法案)和《2012年医疗器械条例》为基础。这些法规由若干通知和指导文件加以补充。
泰国
● 医疗器械和体外诊断(IVDs)由泰国食品和药物管理局(FDA)下属的医疗器械控制司(MDCD)监管。监管框架以《医疗器械法》B.E. 2551(A.D. 2008)、《医疗器械法》(第二版)B.E. 2562(A.D. 2019)以及部长条例和通告为基础。泰国的监管体系目前正在从以前的法规(《医疗器械法》B.E. 2531)过渡到与东南亚国家联盟(ASEAN)医疗器械指令(AMDD)更好地接轨。
新加坡
● 在新加坡,医疗器械和IVDs由卫生科学局(HSA)的医疗器械处监管。IVDs被认为是医疗设备的一个子集。
● 监管框架以《2007年健康产品法》和《2010年健康产品(医疗设备)条例》为基础, 并辅以广泛的指导。监管体系基于东南亚国家联盟(ASEAN)的协调准则和东盟医疗器械指令(AMDD),后者是国际医疗器械监管机构论坛(IMDRF)建议的衍生。
● 所有B类、C类和D类设备都必须进行设备注册。
澳大利亚
● 澳大利亚卫生部下属的治疗用品管理局(TGA)负责澳大利亚的医疗器械监管。IVDs作为医疗设备的一个子集被监管。1989年《治疗用品法》(以下简称 "法案")及随后的《1990年治疗用品条例》和《2002年治疗用品(医疗器械)条例》构成了医疗器械(包括IVDs)监管框架的基础。
新西兰
● 新西兰卫生部(MoH)下属的药品和医疗器械安全局(Medsafe)负责监管医疗器械(包括IVDs)。医疗器械的法律框架基于《1981年药品法》、《1984年药品条例》和《2003年药品(医疗设备数据库)条例》。在Medsafe网站和WAND(网络辅助设备通知)的用户指南中都有相关指导。
Question 4:亚太五国可以独立注册吗? 无CE或FDA的情况下,注册流程有什么不同? 有其他国家的证书可以加快日程吗? 是否需要原产国的注册信息?
马来西亚
● 原产国:原产国注册并不是马来西亚注册的先决条件
● 参考国:全球医疗器械协调工作组GHTF
泰国
● 原产国:无
● 参考国:新加坡HSA
新加坡
● 原产国:无
● 参考国:全球医疗器械协调工作组GHTF
澳大利亚
● 原产国:无
● 参考国:欧盟CE
新西兰
● 原产国:原产国注册并不是新西兰注册的先决条件
● 参考国:澳大利亚TGA
Question 5:亚太五国在产品进口方面需要什么资料? 有什么特殊的要求? 会遇到什么问题?
马来西亚
● 向MDA递交的技术文件基于Appendix 1 of the Third Schedule of the Medical Device Regulations 2012
● 其中包括以下内容:
◆ 执行摘要;
◆ 对于医疗设备。基本原则核对表(MDA/GD/0008的附录A);
◆ 对于IVDs:基本原则核对表(MDA/GD/0004的附录A);
◆ 医疗器械的描述;
◆ 设计核查和验证文件的摘要;
◆ 临床证据 ;
◆ 标签(设备标签、使用说明(IFU)、广告);
◆ 风险分析;
◆ 制造商信息(包括制造商和合同制造商的质量证书)。
泰国
● 泰国FDA要求技术文件采用东盟CDST格式。CSDT格式要求在AMDD的附件4中描述。
新加坡
● 提交给HSA的文件必须是英文的。
● A类技术文件:虽然A类设备不需要在上市前提交档案,但制造商仍然必须拥有证明符合基本原则所需的技术文件。
● B、C、D类技术文件:HSA提交的档案是基于东盟通用提交档案格式(CSDT)--类似于IMDRF的技术文件摘要(STED)格式)。
澳大利亚
● 证明符合澳大利亚法规的技术文件应编制成IMDRF技术文件摘要(STED)格式或目录格式的技术文件文件,而欧盟的技术文件文件/设计档案通常能满足澳大利亚的大部分要求,并有一些补充。
新西兰
● 将设备列入WAND数据库的要求相对较低,在Medsafe网站和WAND用户指南中都有概述。尽管必须确定一个GMDN代码,并询问有关设备类型和设计的问题,但提交的文件并不要求。
Question 6:亚太五国对于上市后监管有什么要求? 进口商或者分销商在方面如何协助,他们有哪些职责范围?
马来西亚
● 根据MDA/GD/0031,制造商必须拥有一个PMS系统以确保持续符合基本原则。对于B-D类器械,CAB审查将包括确认PMS系统已经建立和实施。必须包括以下过程:
◆ 投诉处理;
◆ 分销记录;
◆ 强制性问题/不良事件报告;
◆ 现场纠正行动;
◆ 召回。
● 此外,医护人员和患者可以提交关于医疗器械遇到的问题的报告,然后由MDA根据事件的严重性和/或未来伤害的可能性进行评估。
● 机构应制定符合 MDA/GD/0011《投诉处理》基本要求的投诉处理程序。机构还应保存至少包括 MDA/GD/0012《分销记录》中所述信息的分销记录。
泰国
● 泰国FDA和各省当局对在泰国销售的医疗器械和IVDs以及医疗器械广告进行市场后监督。制造商和代表/经销商必须做好准备,按照医疗器械监管人员的要求提供记录。
新加坡
● 健康产品(医疗设备)条例2010 第八部分对上市后的义务进行了规定。制造商必须保持其设备的制造和批量测试的记录。此外,制造商、进口商、批发商和注册者必须保持条例中规定的器械供应记录。这些记录必须在器械的预计使用寿命内或从器械供应之日起两年内保存。GN-06,《医疗设备分销记录指南》提供了这方面的指导。
澳大利亚
● 包括警戒在内的上市后义务在ARGMD中有详细阐述。
● 澳大利亚有一个高度发达和严格执行的上市后制度,其中包括以下内容:
◆ 报告已导致或可能导致严重伤害/疾病或死亡的国内不良事件(通过保荐人,以电子方式);
◆ 报告全球召回事件(通过主办方);
◆ 采取纠正措施,包括召回或公开通知;
◆ 保持分销记录(IIb级植入式/III级10年;所有其他5年);
◆ 保存器械记录,从最后一个器械的生产日期算起,至少保存5年。
新西兰
● Medsafe可能采取的其他行动包括:
◆ 通过Medsafe网站上的文章和其他出版物向卫生专业人员和消费者提供信息;
◆ 要求对产品或标签进行改进;
◆ 要求赞助商/制造商开展额外的用户教育;
◆ 继续监测市场上的趋势。
关于泰国的,旧规证书已经过期,新规续证已经提交一段时间还在审核,这期间能否正常进口?
● 泰国注册证书Renew在根据《医疗器械法》,BE 2551 (AD 2008)的第一节和医疗器械法,BE 2562 (AD 2019)第21部分中进行了说明。希望renew注册的申请人必须在注册期满前提交续展申请。提交renew申请并支付Renewal费用后,只要泰国 TFDA 批准renewal申请,医疗器械或 IVD 就可以继续上市。renewal费用和原始注册费相同。
● 如果注册证书过期,申请人有1个月的时间申请renewal,需要提交一份免责声明,说明未在到期日之前提交renewal申请的原因,以及renewal费用。注册证书过期超过1个月的,申请人需重新提交注册申请。
在五国注册中,对于短暂侵入人体的器械,如生物相容性报告之前检测了细胞毒、皮内反应,过敏,后面需要补充急毒和热原吗?法规更新了,需要补充测试,还是需要书写说明此次更新的内容不影响测试内容和结果?
● 相关注册如果使用到参考国注册,则需要考虑使用CE,那么CE有对应的标准要求,如果OJ(Official Journal),那么就需要被更新。
如果安规EMC报告中一些较为极端偶尔出现的检测项目达不到条件,是否可以写说明描述相关的情况呢?
● 这个需要第三方检测机构出具报告,如果是Fail的,则不符合相关要求,如果自我宣称,则企业需要对最后的文件进行负责,因为企业永远是最大的责任主体。
针对通过CE认证在澳洲、马来西业取得注册的产品,如果产品的MDR证书更新了,是否需要做变更或者是重新注册?
● 需要及时和当地代表去的沟通,并进行相关产品的更新。
据悉马来西亚目前已出台新政策:同一产品允许多个代理商注册,请介绍下该政策。
● 马来西亚医疗器械管理局官网(MDA):
https://portal.mda.gov.my/announcement/638-pembatalan-surat-pekeliling-pihak-berkuasa-peranti-perubatan-pbpp-bil-1-2014.html
以上就是本期亚太地区多国医疗器械产品注册工作相关内容的问题答疑,希望能实际有效解决大家的问题!