一、杂质的分类与基本概念
按理化性质可分为以下几类:
包括工艺中引入的杂质和降解产物,可能是已知的或者未知的,通常称为有关物质,有关物质又可分为工艺杂质与降解杂质,工艺杂质主要为工艺过程中引入的杂质,包括起始物料、反应物、中间体、副产物、试剂、催化剂等;降解杂质主要由药物降解产生,如水解、氧化、高温、开环、聚合等,与药物的结构特征密切相关。
2)无机杂质:
在原料药及制剂生产过程中带入的无机物,通常是已知的。
3)残留溶剂:
是指在原料药及制剂生产过程中使用的有机溶剂的残留,一般具有已知毒性。
二、杂质产生的主要因素
1)制备过程产生
工艺、辅料等不同产生不同杂质,如盐酸普鲁卡因注射剂高温灭菌过程可能水解为对氨基苯甲酸等,所以在中国药典中原料药不要求该项检查,而注射剂中要求该项检查。
如苯磺酸氨氯地平片处方中采用乳糖为辅料,并存在湿法制粒及粉末直接压片2种生产工艺,由于苯磺酸氨氯地平结构中含有伯氨基,可与乳糖的羰基缩合发生褐变反应,生成氨氯地平乳糖加合物,贮存期片剂的性状可由白色渐变至浅褐色。
2)贮藏过程产生
因保管或贮藏因素、或外界环境影响、或微生物作用可能发生水解、氧化、异构体、聚合、潮解、霉变等变化而产生杂质。
如酯、内酯、酰胺、环酰胺及苷类药物吸湿易水解,如阿托品可水解成莨菪醇和消旋莨菪酸。尤其是酸、碱性条件下或温度高时。
具有酚羟基、巯基、亚硝基、醛基及长链共轭双键等结构的药物,在空气中易氧化,使药物降效、失效,甚至产生毒性。如乙醚在日光、空气及水分等作用下易氧化分解为醛及有毒的过氧化物。
3)药物本身的结构因素
如降血糖药那格列奈,含反式4-异丙基环已酸与4-苯丙氨酸键合的结构基元,若其中的4-异丙基环已酸为顺式或苯丙氨酸为L-型则无活性或活性达不到临床疗效。双羟萘酸噻嘧啶顺式体的驱虫效果仅为反式体的1/60。
4)物料引入
如含乳糖的辅料与主成分含-NH2结构,常常产生乳糖加合物杂质;抗尿失禁药盐酸度洛西汀中2个杂质是因API分别与辅料中的少量琥珀酸和苯二酸反应生成。
5)设备、包材等引入
设备中引入的杂质常见的为元素杂质,但是一些特殊的药物在接触不锈钢后会产生降解杂质,如苯磺酸氨氯地平在溶出试验过程中采用不锈钢的取样针和搅拌桨就容易使API降解,产生新的杂质。
同时包材对于液体制剂质量的影响较大,不同来源的包材在与液体制剂接触过程中易使API降解或者直接引入API中没有的杂质,包材上的油墨也可能迁移至药液中,产生新的杂质,所以液体制剂对包材引入的杂质研究尤为重要。
基于杂质的产生途径,我们可以从以下几方面,对药品的杂质谱进行充分分析:
三、杂质谱分析
1)基于合成路线进行分析
原料药的选择对于仿制药来说至关重要,直接影响到了药品的质量,我们应该学会从原料药的合成路线出发,对原料药中可能引入的杂质进行充分分析,示例如下:
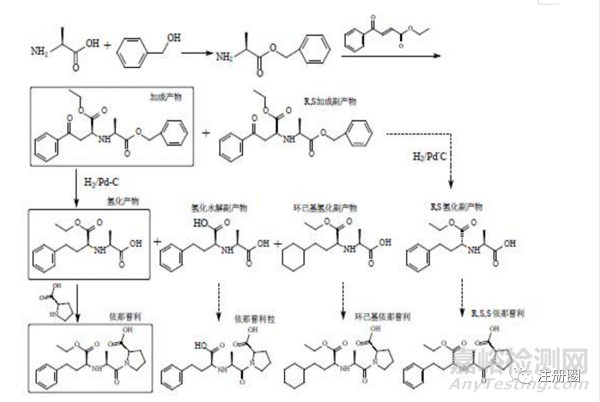
以马来酸依那普利为例进行说明,简略合成路线见下图,以L-丙氨酸为原料,经酯化、加成、催化氢化、成酰胺等多步反应制备马来酸依那普利原料药。在成品的合成路线中会产生副产物、降解产物等,我们可以通过原料药的杂质控制策略了解到更多的杂质信息,进一步评估由原料药引入的杂质。
2)基于官方机构或各国药典进行分析
仿制药的质量标准通常能在各国药典或官方机构中查询到,我们可以通过药典对于杂质的控制进一步了解仿制药的杂质情况。可进行如下汇总:
如某品种BP药典中规定了杂质A、B、C、D、E、F、G、H等8个杂质,其中原料药中均将这8个杂质作为特定杂质控制,制剂药仅控制了杂质1与杂质E,由此可推测杂质A、B、C、D、F、G、H为工艺杂质;杂质1为制剂中的降解杂质;杂质E既是工艺杂质也是降解杂质。ChP药典中仅规定了杂质E,其他各国药典暂无收录。
由上信息收集可知,BP药典中8个杂质基本涵盖了本品在常用制备工艺条件下可能出现的杂质,也为我们后续制剂杂质的研究提供重要的参考依据。
上述信息收集完后,需通过试验对仿制药的杂质谱进行实际考察,主要考察的内容详见“四、仿制药中杂质的研究”。
四、仿制药中杂质的研究
1)分析方法的确定
根据原料药引入的杂质与药典收录的杂质,配制杂质定位与混合杂质分离度溶液进行测定方法的考察,保证方法有一定的检出能力与灵敏度,确定方法后进行制剂的杂质谱研究。
如EP药典中收录的某品种,该测定方法基本能保证各杂质有效检出且分离度良好。
2)制剂中所含杂质的确定
测定多批次的制剂,根据杂质定位确定制剂中所含的杂质种类。
3)制剂中降解杂质的研究
工艺杂质一般由原料药引入,在制剂中无增长趋势,所以制剂中一般不对工艺杂质进行控制,降解杂质为制剂主要的研究对象。了解降解杂质的降解途径对制剂工艺、贮存与运输等有着重要的意义,一般从以下方面进行研究:
强制降解实验(光、酸、碱、氧化、高温)
影响因素(光、湿、热)
稳定性考察(加速、长期)
对试验过程中产生的杂质进行信息收集,如下表:
备注:*表示产生的有机杂质。#原料药内控特定有机杂质指标准中规定要检测并有特定的认可标准的有机杂质;原料药内控非特定有机杂质指标准中其限度在总认可标准中控制而不单独控制的有机杂质(即通过工艺路线推断及各国药典收载,但未按特定有机杂质收载至质量标准的一类有机杂质)。
由上表可知,药典标准收录了杂质A~L,原料药中将杂质A~F作为特定杂质控制,其他均按未知单杂控制,由此可推断杂质G~L在原料药中降解程度很小或者根据合成路线不会产生这些杂质;再根据制剂的稳定性研究情况,可知杂质B、C、E、K为降解杂质,其中杂质B、C、E在原料中也作为特定杂质控制,推测为API自身结构产生的降解;而杂质K在原料药中作为非特定杂质控制,而制剂中存在较明显的降解,推测为API在辅料作用下或者制剂工艺过程中产生。综上分析,制剂中将杂质A~F与杂质K进行定量研究,并将杂质B、C、E、K作为特定杂质控制,其他均作为未知单杂控制。
4)制剂中杂质限度的制定
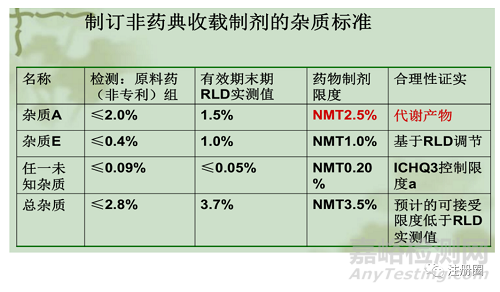
仿制药的杂质限度一般根据各国药典标准或参比制剂的质量进行制定,示例如下: