稳定性相关缺陷和频率
药物稳定性研究是一门成熟的学科,当然也是一项重要的监管要求,近年来许多FDA 483中提到了稳定性计划的缺陷。表1概述了近年来与稳定性相关的典型缺陷和出现频率。
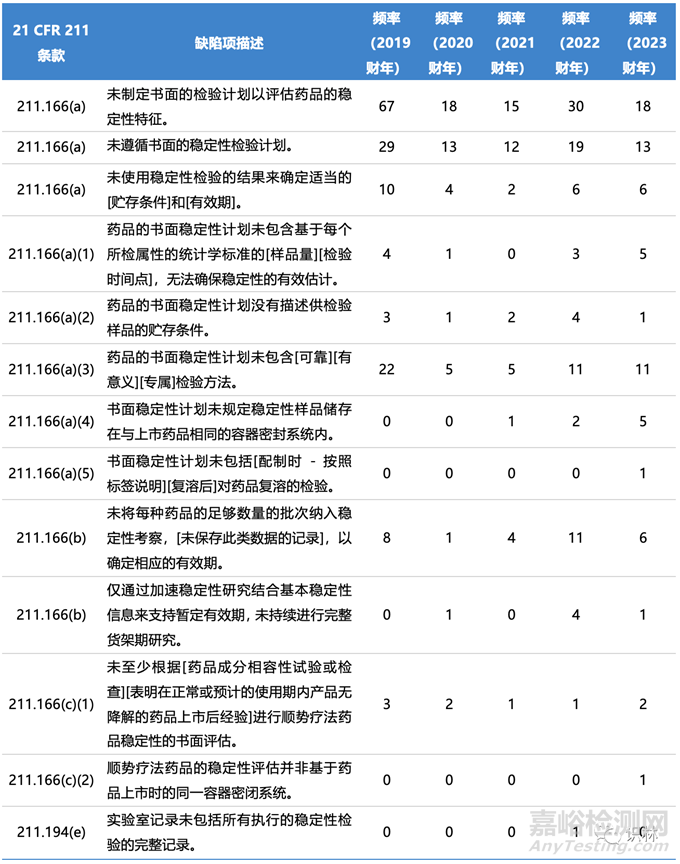
表1. FDA近年来483表涉及稳定性的缺陷项和出现频率
中美检查文件中对稳定性的关注点
除法规要求外,中美监管机构/组织也制定了相应的检查工作程序或合规手册,明确了稳定性的检查范围、要求及风险的判定原则,并列举了稳定性相关的检查缺陷,见表2。
表2. 中美现场检查程序文件中稳定性相关要求和缺陷
稳定性相关FDA警告信案例
20210127 Allay Pharmaceuticals, LLC 美国 FDF
3. 企业未能建立并遵守旨在评估药品稳定性特征的充分的书面检验程序,并使用稳定性试验结果判定适当的贮存条件和有效期(21 CFR 211.166(a))。
企业的稳定性计划是不充分的。企业使用新供应商的API生产的商业规模验证批没有充分的长期稳定性数据支持。此外,企业未能通过始终如一地执行崩解检验来遵守稳定性检验方案。使用新API供应商的验证批次是在2019年5月16日至6月25日生产的,但直到2020年5月1日(差不多一年后)才被纳入长期稳定性研究。
稳定性数据对于确保产品在其货架期内保持鉴别、规格、质量、纯度和安全性至关重要。
在回复中,企业表示已启动了针对稳定性检验缺失的偏差调查,并且这些缺陷不会再次发生。还将聘请稳定性协调员来监督稳定性计划。
企业的回复不充分,因为没有提供临时措施来解决在售产品缺乏长期稳定性数据的问题。
在对此函的回复中,请提供以下内容:
一份综合独立评估以及CAPA计划,以确保稳定性计划的充分性。整改后的计划应包括但不限于:
具有稳定指示性的方法;
每种药品允许分销前在其容器密封系统中的稳定性研究;
持续计划,每年增加每个产品的代表性批次以确定货架期声明是否仍然有效;
每个点(时间点)要检测的具体属性的详细定义;
描述整改后的稳定性计划的以上要素和其他要素的所有规程。