2022年11月30日,NMPA发布“关于实施药品注册申请电子申报的公告(2022年第110号);公告指出,自2023年1月1日起,申请人提交的国家药监局审评审批药品注册申请以及审评过程中补充资料等,调整为以电子形式提交申报资料,申请人无需提交纸质申报资料,这意味着中国药品注册全面进入电子化;那么我们来梳理下电子通用技术文档(eCTD)的发展史,各国实施eCTD进展,以及我国药品注册电子化或eCTD实施进展;
电子通用技术文档(eCTD)的发展史
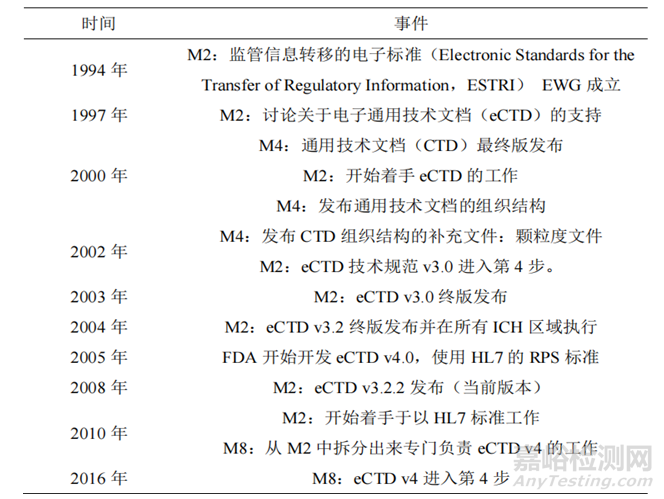
随着经济全球化,各国贸易往来频繁,药品也成为流通大环境的一员;而各国的注册监管法规不同,企业要想在异国申报,需要满足不同监管部门的要求,这将会降低药品上市速度并造成资源浪费;因此美国、欧盟、日本三方成员国组成了国际人用药物注册技术要求协会(ICH),在2000年,ICH开发出一套通用技术文档(CTD),目的是为了提供一种全球统一的药品注册格式,避免多国注册时需要编写不同注册资料。
2003年,ICH M2提出第一版电子通用技术文档eCTD V3.0(技术规范),它是通用技术文档(CTD)的电子版,该技术可进行全程化电子数字管理以及生命周期的管理,引入了MD5 checksum,保证了递交文件的安全性和保密性。在2008年7月16日,eCTD V3.2.2版本开始实施,2015年,为了提高信息的稳健性和灵活性,ICH发布了eCTD v4.0,该版本能够实现信息双通流向功能,V4.0支持所有申请之间的文件使用,例如,一个文件在 IND、NDA中都需要递交时,V4.0只需要递交1次即可,引用其唯一ID来实现该文件重用。2022年,ICH开始大力推广eCTD v4.0。截止目前,大部分国家均处于从eCTD V3.2.2到eCTD v4.0的过渡阶段,其中欧洲、美国、日本这三个国家预计在2026年、2028年、2026年强制执行eCTD v4.0。
表1 eCTD发展史[1]
表2 eCTD v4.0的实施情况[2]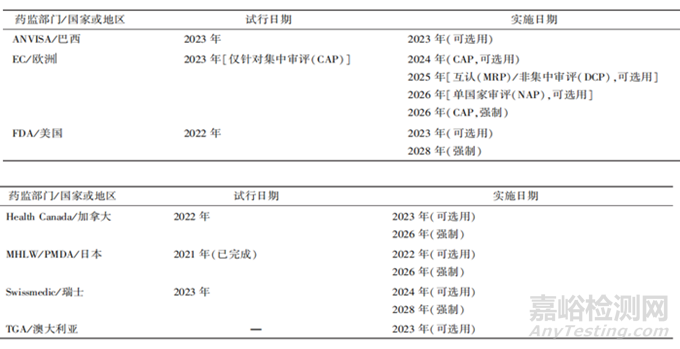
美国实施eCTD进展
在2001年时,美国注册申报方式主要是将CTD格式的注册文档打印下来,采用快递形式进行运送,但其存在的问题是效率低,审核困难;为了提高审评效率,缩短审评周期,2003年,美国FDA开始接受使用eCTD V3.0 进行递交,2005年美国可接受的药品注册申请电子提交格式包括 e-NDA和e-ANDA格式( PDF文件)、e-CTD 格式和NeeS格式等。在2008年1月,FDA规定e-CTD是唯一允许电子提交的格式,当然,在当时FDA的纸质档案也是接受的,也就是说,2008年,FDA进入e-CTD和纸质档案共存过程;这一过程持续了接近9年;在2017年5月15日起,所有的新药申请( NDA) 、仿制药申请( ANDA) 、生物制品申请(BLA) 及其补充申请必须以强制 e-CTD 格式提交;2018年5月5日,FDA要求商业化IND和药品主文件( DMF,3 类DMF除外)申报资料必须以 e-CTD 格式提交,其他所有格式将被拒绝填写,FDA在2018年全面进入e-CTD阶段。
图1美国实行eCTD的发展史[3]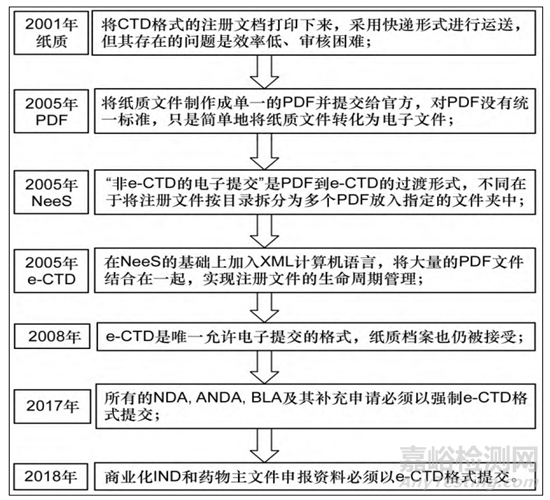
图2 FDA申报流程图[3]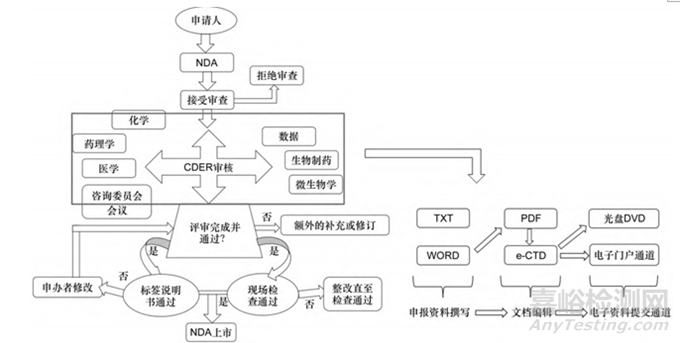
欧盟实施eCTD进展
欧盟在20世纪90年代就提出“电子提交”概念,如由德国开发的 “ DAMOS” 及法国开发的“MANSEV”均为欧盟国家的相关尝试,ICH颁布的eCTD规范于2002年开始在欧盟施行。2007年,由于企业和审评机构对于 eCTD 的使用率低下,有一个变种的格式诞生了,它遵循CTD 的结构但是不支持生命周期管理。它被命名为非 eCTD 电子提交(NeeS)格式,是作为通往全面实行eCTD 之路的过渡形式,欧洲EMA则在2008年7月1日后接受单独的eCTD格式注册文件而不需再提交相关纸质副本,2018年起强制要求所有药品上市都以eCTD格式申报,不要求额外的纸质文件。2010 年7月1日开始,对集中审评的人用药物开始强制要求eCTD 格式。2015年7月1日开始,非集中审评的新的申请开始强制实行eCTD 格式。2017 年1月1日开始,互认可程序的新药申请的提交强制实行eCTD 格式。
日本实施eCTD进展
日本药品和医疗器械局(PMDA)于2004年开始要求使用eCTD,2009年实施eCTDV3.2.2之后,接收到的eCTD 递交数量急剧增加,2015年12月后,日本的大部分新药申请均以eCTD形式提交。
我国药品申报电子化进展
相对于欧盟、美国、日本我国药品申报电子化进展相对较落后,起步较晚,在2004年,我国首次提出综述资料、质量标准、使用说明书和包装标签采用电子提交,这也是我国电子申报的开端,但仅限于部分资料,而且不强制;
2008年05月07日,为了提高审评效率,NMPA发布“关于通过中心网站提交药品注册申报资料电子文档的通知”,这是在《药品注册管理办法(局令28号)》发布后,NMPA启动了过渡期品种的集中审评工作。通知要求:在CDE网站上传docx格式的文件,适用范围:2007年10月1日之前已受理但尚未完成审评,且之前未进行过电子提交的注册申请,2007年10月1日之后已受理,但此前未进行过电子提交的注册申请。提交内容包括质量标准(草案)、药品说明书、立题目的与依据、对主要研究结果总结与评价、药学研究综述、药理毒理研究资料综述、综述临床试验研究资料综述等资料;该通知的发布打开了我国药品申报电子提交的开端。
2013年04月药审中心将试点开展药品注册申报资料电子递交工作;2017年11月国家局发布“关于调整原料药、药用辅料和药包材审评审批事项的公告(2017年第146号)”公告提出:原料药、药用辅料和药包材企业在药审中心门户网站“申请人之窗”填写品种基本信息后,将登记资料以光盘形式提交至药审中心;这是我国首次提出以光盘形式提交资料,但目的仅用于现场核查检验;
2019年5月6日,CDE发布“关于提交药品注册检查检验用申报资料光盘的通知”,根据该通知的要求,应在受理后10日内提交光盘,主要目的是方便现场检查和检验,而非用于受理或审查;
2020年4月30日,CDE发布“公开征求《化学原料药受理审查指南(征求意见稿)》意见的通知”;2022年2月9日,CDE发布“关于再次公开征求《化学原料药受理审查指南(试行)(征求意见稿)》意见的通知”,本通知中的受理审查指南是对“2017年第146号”公告的落实,在“2017年第146号”公告中要求原辅包以光盘形式递交申报资料,虽然至今尚未转正,但业界已经按照最新征求意见的受理审查指南,开展原料药的申报。
2022年1月29日,CDE发布“关于疫情期间调整受理工作方式及接收申报资料要求的通知”,为了控制日益增多的纸质资料邮包带来的疫情传播风险,CDE发布了该通知。虽然本次通告并非eCTD申报,但其提出了基于光盘资料进行受理审查的理念,也是我国药品申报电子化进程中的重要节点,不过这次要求受理后5个工作日提交纸质资料,也就是说这期间还是要求电子光盘+纸质资料。
2022年11月30日,NMPA发布“关于实施药品注册申请电子申报的公告(2022年第110号公告;公告指出,自2023年1月1日起,申请人提交的国家药监局审评审批药品注册申请以及审评过程中补充资料等,调整为以电子形式提交申报资料,申请人无需提交纸质申报资料,申请人采用药品电子通用技术文档(eCTD)进行申报的,无需再提交纸质申报资料,这意味着中国全面进入电子申报时代,在这期间eCTD格式也是被认可。
图3 2008年药品审评中心电子提交系统[4]
图4 我国电子申报发展史(注:红色文字为eCTD相关的法规,其他为电子注册申报相关)
综上所述,中国的药品注册电子化主要经历如下,①2004年部分资料电子化(综述资料、质量标准等资料电子化);②2008年部分资料电子化(在2004年电子化的扩充)+纸质资料提交;③2017年,申报资料光盘提交主要用于现场核查,而审查及申报资料与2008年(部分电子资料+纸质资料)相同;④2020年,原辅料申报采用电子申报(无纸质),不过这次是征求意见稿,大部分企业按照征求意见稿实行原料药申报;⑤2022年,因为疫情申报资料电子光盘提交+纸质资料;⑥2022年,中国进入全面电子申报(光盘形式),无需提交纸质申报。
我国eCTD实施进展
2017年03月15日,药审中心开始加快推进eCTD项目建设,2017年05月30日药品审评中心发布关于征求《药品电子通用技术文档结构(征求意见稿)》和《化学仿制药电子通用技术文档申报指导原则(征求意见稿)》意见的通知,2017年10月再次发布关于药品电子通用技术文档结构的征求意见稿,直到2021年9月30日正式发出公告,国家药监局关于实施药品电子通用技术文档申报的公告(2021年第119号),实现药品注册申请的电子申报,提升“互联网+药品监管”应用服务水平,国家药品监督管理局全面开展和推进了药品电子通用技术文档(eCTD)申报相关工作,实施范围化学药品注册分类1类、5.1类,以及治疗用生物制品1类和预防用生物制品1类的上市许可申请,可按照eCTD进行申报,申请人应按照eCTD技术文件(附件1-4)要求准备和提交eCTD申报资料光盘,并在eCTD注册申请新报资料受理后5个工作日内,提交纸质;这是我国正式eCTD申报的开端,不过,这次eCTD并不够彻底,因为提交eCTD后还需要提交纸质资料,而且仅针对部分,提交的介质为光盘,而非网关至网关传输;并且非强制性要求。
由此可见,我国对于eCTD的目前处于选用阶段,仅仅是小范围实施,主要还是以电子注册为准,eCTD不强求,估计还有很多因素需要考虑,特别是企业的响应程度。
总结:
从eCTD的发展史看,我们得知,eCTD的版本在不断的升级,管理和使用也是变得越来越方便;从各国eCTD实施情况看,美国、欧洲、日本实施eCTD较早,大部分都经历过纸质资料申报阶段、纸质资料+电子申报共存的过渡阶段、电子申报(格式多样)、最后进入强制执行eCTD阶段;而我国目前正进入电子申报+eCTD共存阶段,相信不久将来,我国可能也会与国际接轨,完全进入eCTD阶段。
以上是基于当前认知水平进行分析总结,如有错误,欢迎大家留言指正。
参考文献:
[1]吕婷等 美国和欧盟药品申报电子提交的对比研究及对中国实施的启示 2018 - 上海交通大学;
[2]陈 华等 我国正式实施 eCTD后药品生产企业的注册申报应对策略 chinese journal of new drugs 2023 32(12)
[3]丁慧颖等 美国基于电子通用技术文件格式的药品注册申报制度研究及启示 Chinese Journal of New Drugs 2023,32(21)
[4]NMPA官网
[5]李东昂等 我国药品注册申报资料实施eCTD格式的策略研究 中国制药装备·2018 年3 月·第 3 辑