摘要
儿童药品的供应保障是儿童医疗保健的重要一环,然而我国的儿童药品的供应保障却不能满足市场需求。国际上欧美等国二十多年来已经建立了一套行之有效的儿童用药法律法规及配套激励措施,极大的促进了儿童医疗保健事业的发展。本文通过对比分析欧美促进儿科药品研发的关键工具-儿科药品研发计划(PSP和PIP)的内容及审评审批流程,建议监管当局在儿童药品政策法规的规制定和调整时引入儿科研究计划并设立儿科药品审评委员会,从而引导我国制药企业研发理念的转变,在药品研发早期就将儿童纳入整体研发策略中。
关键词:儿童用药;儿科药品研究计划(PSP);儿科药品调查计划(PIP);审评审批
引言
儿童医疗保健是儿童健康发展的重要保障[1],保障儿童用药的安全性和可及性是确保儿童健康发展的重要一环。ICH E11(R1)[2](用于儿科药物的临床研究)将儿童患者按年龄分为足月新生儿(0-27天)、婴幼儿(28天-23个月)、儿童(2-11周岁)、青少年(12到16-18周岁)四个亚群。在我国儿童通常是指0-14岁的人群,巨大的人口基数[3]及高患病率和就诊率[4]引爆儿童用药市场需求。然而,我国儿童用药的供给保障却不容乐观,存在品种短缺(普药多,特药少)、剂型与规格不匹配儿童生理需求等困境。相关法律法规指导原则不完善、儿童药品研发对辅料和剂型有特殊要求(如适口性)、不同年龄段儿童生理病理的差异和临床试验在伦理及安全方面要求高以及儿童患者难招募易脱落等问题导致儿童药品研发成本高、周期长[5],使许多有意愿生产儿童药品的企业望而却步,由此进一步造成我国儿童用药领域超说明书用药现象严重[1],给儿童的身心健康带来极大风险。
儿童药品市场相对于成人药品市场来说是“小市场”,由此导致儿童用药的研发往往是成人药品研发的附属,即儿童药品的开发一般是由成人药品开发所驱动,而不是由儿童的健康需求驱动。此外儿童药品的研发不应局限于儿童专用药品,更多的是应鼓励满足儿童用药需求产品的儿科研究,最终完善药品说明书的儿童用药信息,从而降低超说明书用药的风险,保障儿童身心健康[6]。
我国针对儿童用药的政策起步较晚,目前还没有针对儿童用药的专门立法,相关法律法规还停留在部门层面,不具备强制执行力,系统解决儿童用药的安全可及问题亟需上位法的建立健全。相比之下,国际上欧美等国对儿童用药的立法工作已积累了二十多年的经验,逐步建立了一套行之有效的儿童用药法律法规及配套激励措施[1],极大的促进了儿童医疗保健事业的发展。
本文主要对欧美促进儿科药品研发的主要工具-儿科药品研发计划的内容及审评审批流程进行对比,以期为监管部门在制定促进我国儿童药品研发政策时提供借鉴。
一、法规背景
美国通过《儿科研究平等法案》(Pediatric Research Equity Act ,PREA)引入了儿科药品研究计划(Pediatric Study Plan,PSP),通过《最佳儿童药品法案》(Best Pharmaceuticals for Children,BPCA)引入了书面请求(Written Request,WR),通过这两个工具,采用强制与激励相结合的方法引导企业进行儿科研发。欧盟通过儿科管理条例(Regulation(EC)No 1901/2006)引入儿科药品调查计划(Pediatric Investgation Plan,PIP),采用强制与激励加帮扶的方法引导企业进行儿科研发。
1.1 美国
在美国儿童是指0-17岁的人群[7]。美国主要通过BPCA和PREA两大支柱性法案促进儿科药品的研究。
BPCA于2002年1月4日正式生效实施,允许美国食品药品监督管理局(Food and Drug Administration,FDA)对认为可能产生儿科获益药品的申请人发放WR,以请求申请人进行儿科研究,WR中会列出拟研究的类型、适应症、年龄组、研究终点、评估时间、入组标准、药品信息、剂型和给药途径、研究方案、安全性信息、统计信息、标签信息、报告格式及时间框架[8]。WR为非强制性要求,但如果申请人严格按照WR在规定的时间内完成了研究并提交了报告,无论研究结果是否积极,FDA都会授予申请人6个月的儿科独占期(pediatric Exclusivity,PE),附加在申请人持有的含有相同活性成分的药品的专利期或者市场独占期后面。申请人也可以提交拟议的儿科研究请求(Proposed Pediatric Study Request,PPSR),主动向FDA申请WR。
2003年12月3日,美国国会通过PREA,除非FDA授予豁免(waiver)或者延迟(deferral),否则要求特定药品或者生物制品的申请或补充申请(如新活性成分,新适应症、新剂型、新给药方案或新给药途径)进行儿科评估(pediatric Assessment)[9],即评估药品或生物制品在所有相关的儿科人群中所声称适应症的安全性和有效性,以支持在相应儿科人群中给药方案的安全和有效。PREA为强制性条款,完成儿科评估是产品获批上市的前提。PREA不具有激励性,但PREA只针对产品所申请适应症在相关儿科人群中的安全和有效性评价;而WR主要针对该产品的活性成分,可能要求在不同儿科人群中进行多个适应症的研究。2012年的《FDA安全与创新法案》(FDASIA)对PREA进行修订,引入PSP,要求特定申请的申请人在提交上市许可申请前需要提交初始PSP(Initial PSP,iPSP)并得到FDA对iPSP的同意。
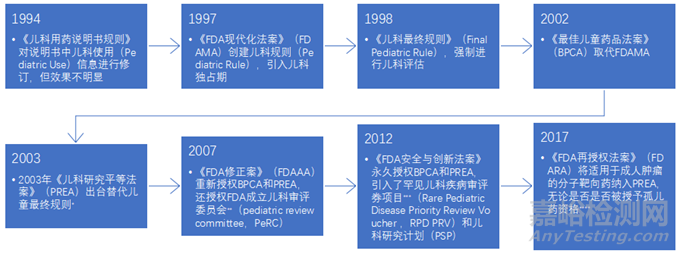
BPCA和PREA有日落条款,2007年被重新授权,2012年被永久授权此后又经历一轮修订(见图1)。
▲图1-BPCA和PREA的立法沿革
*《儿科最终规则》于2002年末被哥伦比亚特区联邦地方法院判定违宪,禁止其实施[10]。PREA继承了大部分《儿科最终规则》中的条款。
**FDA儿科审评委员会(PeRC)主要负责审评PSP、WR、PPSR及相关的豁免和延迟,并向FDA提出建议,其建议没有强制性。
***儿科罕见疾病优先审评券项目(RPD PRV),授权FDA对符合相关儿科罕见疾病标准的申请授予RPD PRV,该券可以转让,可以使使用该券的标准申请(300日)获得优先审评(180日)的资格。
****《FDA再授权法案》(FDAAA)实施前,被认定为孤儿药的产品被排除在PREA的覆盖范围外,即不用进行儿科研究。
1.2 欧盟
欧盟对儿童的定义一般为小于18岁的人群[7]。欧洲药品评价局(EMEA) [2004年改名为欧洲药品管理局(EMA)]自成立起(1995年)就非常重视儿童用药的安全性和有效性。欧盟对儿童用药的正式立法开始于1997年欧盟委员会在EMA组织的一次专家圆桌会议,2000年ICH E11发布,EMA将其纳入期监管指南。欧盟发布儿科监管条例(Pediatric Regulation)公布于2006年6月1日并于2007年1月26日正式生效实施[11]。该条例的目的是确保高质量的儿童药品研究,确保儿童用药品有适宜的剂型以及促进高质量的儿科用药信息的可获得性;同时避免儿童参加不必要的临床试验和成人用药品的批准被延迟[12]。
在儿科用药监管条例的框架内:
引入了PIP,规定新药上市授权申请(Maketing Authorisation Application,MAA)以及新适应症、新剂型、新给药途径的补充申请都要包含达成一致的PIP所涉及的儿科研究结果,以支持该药品在相关儿科人群中的授权和使用,除非该药品被授予豁免或者延迟。
创建了儿科委委员(Pediatric Committee,PDCO)负责对PIP的审评,不同于FDA的PeRC,PDCO的审评意见EMA必须采纳。同FDA一样,无论研究结果是否积极,申请人按照要求完成与PDCO达成一致的PIP的研究后,即可获得6个月专利补充保护延长,或者2年的市场独占期延长(孤儿药)。
此外该条例针对专利到期的药品还提出了新的上市路径-儿科使用上市许可(Pediatric Use Marketing Authorisation,PUMA),并配备10年数据保护期的激励措施,但在EMA 2017年的报告中该条款的实施效果不佳[12]。
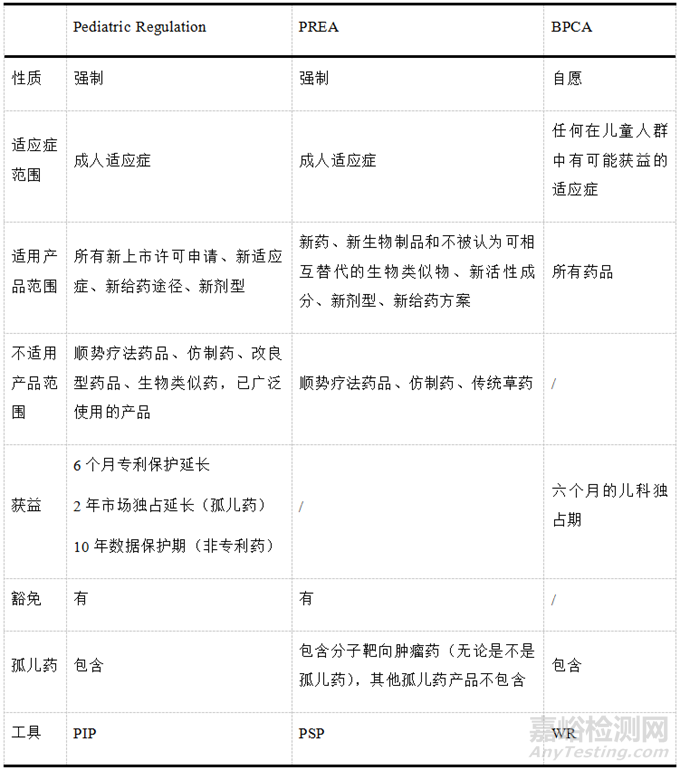
该条例还针对儿科用药信息的公开透明建立了泛欧洲儿科药物研究和临床试验中心网络(Enpr-EMA),并将研究结果录入欧盟临床研究数据库(EudraCT)。还有一系列其他的帮扶措施如针对儿科研究中遇到的问题可以免费向EMA寻求科学建议或方案协助(针对孤儿药)。该条例与BPCA与PREA的异同见表1。
▲表1- BPCA、PREA与Pediatric Regulation之间的异同
二、欧美儿科药品研发计划内容[13-16]
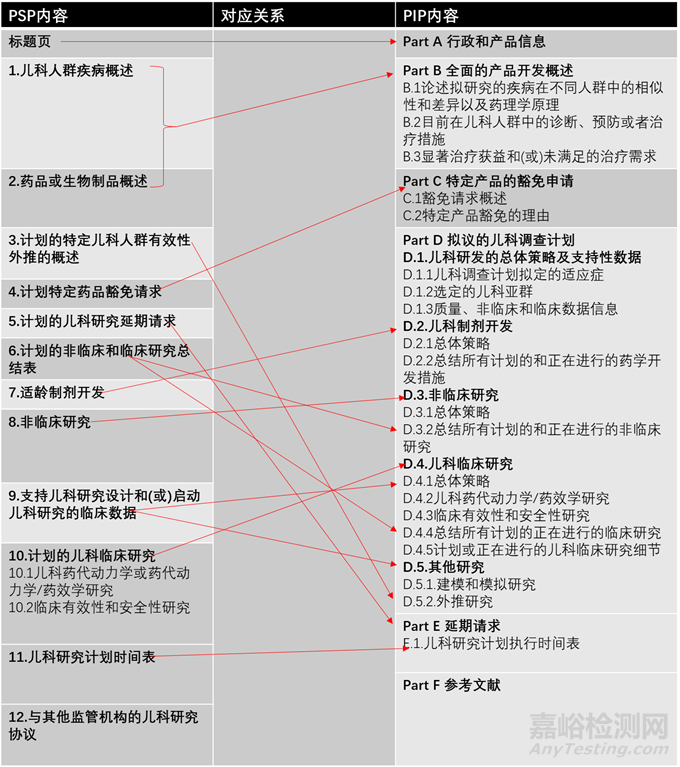
FDA和EMA都要求PSP和PIP是简洁独立的文件,PSP一般为12-60页,PIP不超过40页。PSP和PIP内容上基本相似,但在层级结构上有所不同(见图2)。
▲图2-PSP和PIP内容及其对应关系
2.1 行政和产品信息
PSP要求在标题页中提供行政和产品信息,包括药品专利名和通用名、 NDA/BLA/IND号、药物类别、适应症、研究计划简介等;相对于PSP,PIP的要求较多包括申请人信息,活性成分名称、产品类别、产品细节(剂型、规格和给药途径等)、产品的上市授权状态、从监管机构获得的在相关儿科人群开发的建议、在欧盟是否为孤儿药、计划提交MAA的日期、研究计划简介等。
2.2 疾病概况及产品开发背景
PSP要求分开论述儿科疾病的概况及产品开发信息(第1和第2部分),而PIP将疾病信息整合到产品开发信息中在Part B部分统一论述。PSP和PIP都要求针对拟研究疾病的病理学生理学,疾病的严重程度及疾病在成人与儿童人群中的相似性与差异,目前的诊断、预防和治疗措施,儿科人群中的发病率以及该产品在儿科人群中潜在的治疗获益进行讨论。
2.3 对有效性外推研究的概述
从参考人群(如成人或大龄儿童)向目标人群(如儿童或低领儿童)的有效性外推,可以在避免儿童参加不必要的临床试验的同时获得儿科用药信息。PSP要求在第3部分概述外推计划,而PIP要求在Part D.5部分概述。若有外推计划则需列出疾病在参考人群与目标人群相似性的支持性信息如疾病的发病机理,诊断标准,病理生理学、组织病理学、病理生物学特征,暴露量-反应关系及建模和模拟研究等。
2.4 豁免申请
PSP要求在第4部门提出豁免申请,而PIP要求在Part C 部分提出。FDA与EMA对豁免申请的理由都做出了具体的规定,只有满足这些规定才能申请豁免,同时要提供支持性信息。
FDA规定可以申请豁免的标准是:1)必要的研究是不可能或者高度不可行的(如患者数量极少);2)有证据显示该产品在全部或者部分儿科患者中是无效或者不安全的;3)该产品与现存的治疗方法相比不具有显著的治疗获益且不可能用于实质数量的儿童患者;4)开发适龄的儿科剂型是不可能的。EMA规定可以申请儿科豁免的标准是:1)产品在儿童患者中无效或不安全;2)该药品打算治疗的疾病只发生在成人中;3)与现存的儿科患者可获得的治疗相比,该药品没有显著的治疗获益。豁免可以是部分的,即只豁免部分儿科人群的研究。如果该药品针对的疾病只发生在部分儿科患者中或者针对部分儿科患者开发适宜的剂型是不可能的。
药品开发是个漫长的过程,随着信息的积累,豁免可能会被推翻,豁免申请在PSP和PIP被批准时都只代表监管当局在当时的决定,正式的豁免申请在做出上市批准决定时会被正式授予。
2.5延期申请
PSP要求在第5部分提出延期申请,而PIP要求在Part E 部分提出。同豁免一样延期申请也有特定标准,PSP:1)如果在儿科研究完成前该药品已经准备好用于成人;2)儿科研究应该被延期直到获得足够的安全和有效性信息;3)其他合适的理由(如适龄剂型的开发未完成)。PIP:1)科学技术理由;2)与公共健康相关的理由;3)开始儿科研究之前进行成人研究是合适的;4)儿科研究花费的时间会比成人研究长很多。在两个地区申请延期的标准是十分相似的,但经济因素都不能成为延期申请的理由。同豁免一样延期申请也只有在批准上市时正式授予。
2.6 列表总结计划的非临床和临床研究
PSP要求在第6部分列表总结所有计划的非临床和临床研究,而PIP则分散在Part D.3非临床研究和D.4临床研究中。
2.7 适龄剂型开发
若目前的剂型不适用于所有的儿科人群,两个地区都要求提供适龄剂型的开发计划。PSP要求在第7部分概述,而PIP分散在Part D.1.3及D.2中。应提供处方、剂型、规格、给药途径和辅料信息及开发策略。PIP还要求所有的质量相关的信息作为独立的文件提供。
2.8 非临床研究
PSP的第8部分与PIP的Part D.1.3和D.3是关于非临床研究的内容。在该部分要总结已有的支持临床开展的非临床数据,评估是否还需要开展额外的非临床研究。若需要开展相关研究应简要描述研究计划:包括研究类型、物种,起始剂量,持续时间,给药途径及结果监测方法。
2.9 已有的支持儿科临床研究设计和启动的临床数据
PSP应在第9部分简要总结已经在成人临床研究中获得的安全性、有效性及药动学和暴露-反应数据,以支持相关儿童人群的研究。如果有用于筛选儿科剂量的建模和模拟研究计划也要包含在该部分中。PIP与之相对应的信息分散在Part D1.3、D4.1、D4.4及D5.1部分。
2.10 临床研究
2.10.1 儿科药代动力学/药效学研究(PK/PD)
PSP要求在第10.1节按照第6部分列出的计划的临床研究表的顺序讨论儿科PK/PD研究。应包含研究类型/研究设计、儿科年龄群、拟用的儿科制剂、剂量范围、研究终点及其论证(如PK参数,PD生物标志物)、支持剂量选择的建模和模拟研究、计划的药物基因组学分析及样本量论证。PIP需要在Part D4.2部分列出研究大纲,并在D4.5部分提供细节。
2.10.2 临床有效性和安全性研究
PSP的第10.2节应按照第6部分列出的计划临床研究表的顺序概述儿科临床安全性和有效性研究。应包括研究类型/研究设计、研究目的、儿科年龄群、入排标准、主要和次要终点、终点评估的时间点、统计学方法等。PIP需要在Part D4.3部分列出研究大纲,并在D4.5部分提供细节,主要概述研究类型、研究设计和控制、主要目的、研究的儿科年龄群、最小样本量、研究用药信息(剂型、剂量、治疗方案及给药途径)、主要终点和次要终点及其评估时间、统计计划等。
2.11 研究计划时间表
PSP的第11部分和PIP的Part E.1要求列出所有计划进行的药学、非临床和临床研究时间点。如方案提交时间、研究开始时间、研究完成时间、最终研究报告提交时间。还要提供预估的上市申请的提交时间。同意PSP和PIP不等同于同意完整的临床研究方案,在临床研究开始前还需要向监管当局提交完整的试验方案。
2.12 与其他监管机构的儿科研究协议
PSP要在第12部分提交与其他监管机构达成的儿科研究协议(如经PDCO审评过的PIP)。PIP虽没有此要求,但要求在Part F提供所有支持上述科学讨论相关的参考文献,在此也可以提交其他地区监管机构的建议,比如FDA的WR。
三、欧美儿科药品研发计划审评审批流程
3.1 PSP审评审批流程
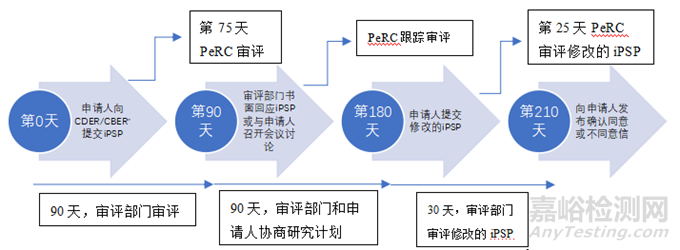
申请人一般应该在Ⅱ期临床结束(EOP2)会议召开后的60天内提交iPSP;如果没有EOP2会议,iPSP应在Ⅲ期临床或Ⅱ/Ⅲ期临床开始前召开会议时提交;若以上都没有申请人应在预计的上市申请提交日期的210天之前提交iPSP[14]。
申请人向FDA相关审评部门提交iPSP后,审评部门有90天的时间进行iPSP的审评并书面回应或者与申请人召开会议讨论。其中PeRC对iPSP的审评应不晚于第75天。接下来申请人有第二个90天去回应审评部门的要求,期间申请人可以与审评部门对研究计划进行协商,PeRC也会跟踪审评,第二个90天结束后申请人必须提交修改的iPSP(Agreed iPSP)。随后,FDA有30天时间审评修改的iPSP,PeRC应不晚于第25天审评修改的iPSP并向申请人发出确认同意或者不同意信。审评审批程序见图2[17]。
*CDER:FDA药品审评与研究中心;CBER:FDA生物制品审评与研究中心
▲图3-PSP审评审批流程及时间线
3.2 PIP审评审批流程[18]
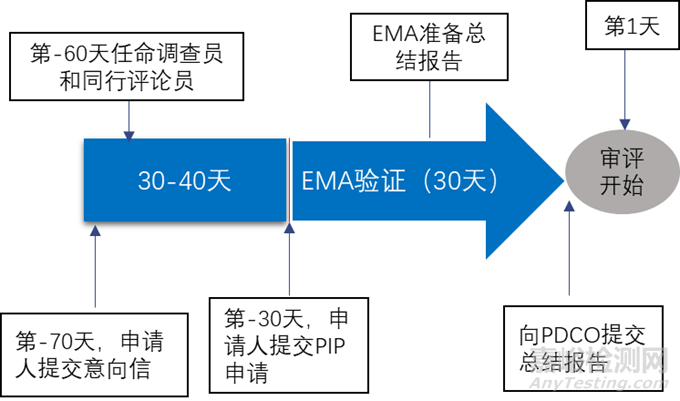
申请人应在不晚于成人药代动力学研究完成的日期提交PIP,一般为Ⅱ期临床开始之前,不应在Ⅲ期临床试验开始后提交。EMA鼓励申请人在人体药代动力学研究完成之前提交PIP,尤其是针对治疗严重的或威胁生命的疾病的药品。
申请人一般需要在正式审评开始前两个月提交意向信,随后EMA会任命一名PDCO (Paediatric Committee) 调查员与一名PDCO同行评论员负责起草总结报告,申请人在提交前可以申请提交前会议。提交申请的时间约为正式审评开始的前一个月,在这一个月内EMA会对PIP进行验证(validation),主要验证信息的正确性和完整性(行政验证),PIP的结构是否正确以及是否有足够的科学信息(监管与科学验证)。验证完成后,EMA汇总并撰写总结报告,并提交给PDCO,审评开始。
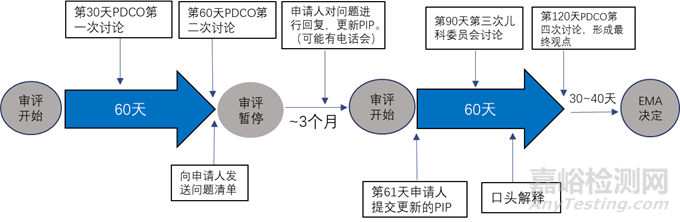
审评开始后的第30天,PDCO对报告进行讨论,并形成总结报告,总结报告中包含PDCO的反馈以及初步观点,该报告会发送其他专家以收集评论,同时也会转发申请人。第60天,PDCO进行第二次讨论,若认为该PIP没有问题则PDCO审评程序结束;若认为PIP中还有未解决的问题则会向申请人发送问题清单,审评暂停。审评暂停期间申请人根据问题清单对PIP进行修改,在此期间可以与PDCO进行电话沟通。申请人提交修改的PIP,审评开始,在第90天,PDCO进行第三次讨论,主要针对申请人提交的修订的PIP,并形成总结报告。如果还有未解决的问题申请人或者PDCO在第三次讨论后可要求口头解释,即申请人直接与整个儿科委员会对话。PDCO的最终观点和最终报告会在第120天的第四次讨论后发给申请人。若申请人对PDCO的最终观点有异议,可以在30天内请求重新评估,随后的10天内EMA会根据PDCO的最终观点和总结报告做出决定。流程图见图4和图5。
▲图4-PIP审评审批流程(验证阶段)
▲图5-PIP审评审批流程(评估阶段)
3.3 审评审批流程对比
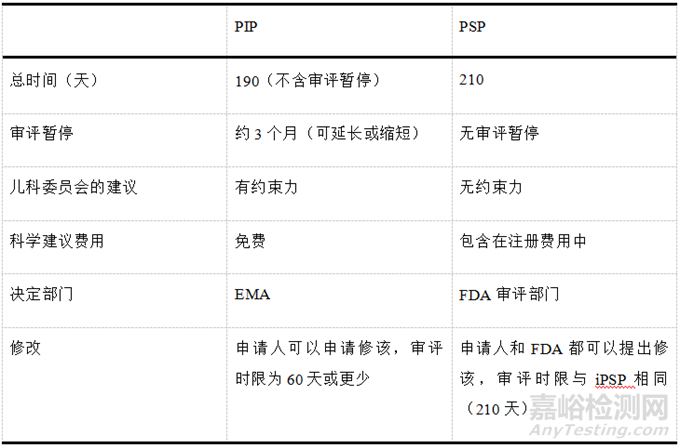
EMA和FDA都鼓励申请人尽早提交儿科研发计划,并鼓励申请人在提交研究计划之前与EMA或FDA沟通,以便更好的将儿科研发计划纳入整体开发策略。儿科临床试验逐渐趋向于多区域多中心临床试验,各区域之间监管要求的协调统一对于加快儿科药品开发,避免儿童参与不必要的临床试验具有重要意义。分析PSP和PIP的审评流程,存在协调统一的可能。因为PIP规定的提交时间比PSP的早,所以可以先提交PIP,在第60天收到PDCO的问题清单后,在随后PIP的审评暂停期间,根据问题清单更新儿科研究计划,向FDA提交iPSP,随后在第90天收到FDA的回馈后,再更新PIP,向PDCO反馈。虽然PIP的审评暂停时限一般为3个月,但审评暂停时限可以根据具体品种延长或缩短,iPSP的审评流程理论上可以在此审评暂停时限内开始,并在此时限内获得FDA的反馈建议,从而规避部分由于欧美对儿童用药临床试验监管要求不同所造成的阻碍。PSP与PIP的审评审批流程对比见表2。
▲表2-PSP与PIP审评审批流程对比
讨论
欧美等国关于儿童临床试验的理念已转变为通过让儿童参与临床试验来保护儿童,制药企业已经在药品开发早期就将儿童人群纳入整体开发策略。据报道EMA已经批准了超过1000个PIP申请[12],根据PERA和BPCA修改说明书中儿童用药相关信息的药品超过900多种[9],极大的促进了儿童医疗保健事业的进步。
我国从2011年发布《中国儿童发展(2011~2020)》以来开始重视儿童用药的供应保障,近年来通过将儿童药品纳入优先审评,设立儿童用药特殊标识以优化审评资源,成立儿童用药专项领导小组和工作小组,完善儿童用药审评标准体系,开展已上市药品说明书中儿童用药信息规范化增补工作等举措[19],此外国家药监局近期发布的《中华人民共和国药品管理法实施条例(修订草案征求意见稿)》提出12个月儿童用药独占期,极大的激励了我国儿童用药的研发工作,2021年全年已有47个儿童药品获批上市[20]。
虽然已经取得了如此的进步,但应意识到我国的儿童用药品种少、剂型少、规格少、特药少的局面还没有完全改善。基于此并结合本文研究内容给监管当局提供以下几点建议:
1)尽早启动儿童用药立法工作:从欧美等国的经验不难看出,强制性的法律要求以及相关激励和帮扶措施是儿科用药领域取得进步的前提条件。我国应尽早启动儿童用药专项立法工作,不仅要涵盖儿童用药品的研发和生产,更要包含流通使用、上市后监测、再评价等药品全生命周期的内容。
2)引入儿科研究计划: 欧美的儿科法律都引入了专门的工具(PSP和PIP)以促进儿童药品研发,同时在其内容和审评审批流程上发布了相关指南以指导企业进行儿科研发。我国可先试点针对某些特定类别的药品(如超说明书用药现象严重的药品),制定相关激励措施(如资金支持和技术指导),在相关指导原则完善的基础上强制相关企业或研究机构提交儿科研究计划,具体要包括产品开发背景、不同儿科年龄组的安全性和有效性评估、豁免和延期申请、外推计划、已有的支持儿科研究信息、药学开发计划、非临床和临床开发计划及其启动和完成的时间点。
3)成立儿科药品审评委员会:欧美都有专门的儿科药品审评委员会(PeRC和PDCO)专门负责对儿科药品研发计划及其豁免和延期申请进行审评并为审批决定提供建议,PeRC还要负责审评WR和PPSR。委员会成员涵盖儿科专家、新生儿专家,还有涉及临床药理、统计、毒理、安全性、化学、法律、伦理方面的专家。我国在引入儿科研究计划的同时可以成立儿科药品审评委员会负责对儿科研究计划的审评以及制定儿童用药审评标准及指导原则,同时为申请人提供科学建议,促进儿童用药监管科学发展。
4)增强国际合作:欧盟、美国、加拿大、日本和澳大利亚等国的监管机构每月会举行电话会[21]讨论收到的关于儿科研发计划的内容,会后还会将会议上达成的共识转告申请人,以降低监管成本,促进儿科研发计划的协调统一,在避免儿童参与不必要的临床试验的同时加快儿童药品开发。我国自2017年加入ICH以来,2018年当选ICH管委会成员,2021年连任管委会成员[22],药品注册监管制度国际化明显,已能够在药品监管的国际舞台上发声。儿童药品研发有趋于多区域多中心的趋势,我国应积极参与儿童用药研发的国际合作,为儿童药品研发的国际协调提供新思路、新方法、新工具,同时鼓励跨国药企在国内研发儿童药品。
5)加强宣传教育促进理念转变:目前国内大众对于儿童参加临床实验的接受度不高,普遍的理念还是通过避免儿童参加临床试验来保护儿童,而非通过促进儿童参加临床试验来保护儿童;除了专门针对儿童的药品,制药公司也未在药品开发早期就将儿童群体纳入药品开发整体策略中,而是在成人适应症批准后才开始考虑儿童的适用性。监管当局应当对大众加强宣传教育,对企业加强沟通交流和培训指导,充分利用各学术团体的影响,促进大众和制药企业理念的转变。
参考文献
[1] 史录文,王晓玲,国家卫生和计划生育委员会儿童用药专家. 中国儿童用药立法研究[M]. 北京:中国协和医科大学出版社, 2017.06.
[2] ICH HARMONISED GUIDELINE. ADDENDUM TO ICH E11: CLINICAL INVESTIGATION OF MEDICINAL PRODUCTS IN THE PEDIATRIC POPULATION[EB/OL]. https://database.ich.org/sites/default/files/E11_R1_Addendum.pdf.
[3] 第七次全国人口普查公报(第五号)[EB/OL]. http://www.stats.gov.cn/tjsj/tjgb/rkpcgb/qgrkpcgb/202106/t20210628_1818824.html.
[4] 生物医药:政策支持下,儿童专用药步入发展快速路-研究报告正文 _ 数据中心 _ 东方财富网[EB/OL]. https://data.eastmoney.com/report/zw_industry.jshtml?infocode=AP202208171577291886.
[5] 许淑红,张绮,张林琦,等. 探讨我国儿科用药的发展现状及政策层面的思考[J]. 中国临床药理学杂志, 2020, 36(12): 1760-1767.
[6] 龚传欢,杨悦,田丽娟. 儿科药物研发激励政策研究_龚传欢[J]. 中国新药杂志, 2022, 31(11): 1042-1047.
[7] Regulatory Aspects of Paediatric Drug Development. — Scendea[EB/OL]. https://www.scendea.com/regulatory-aspects-of-paediatric-drug-development.
[8] Guidance for Industry Qualifying for Pediatric Exclusivity Under Section 505A of the Federal
Food, Drug, and Cosmetic Act[EB/OL].https://fda.report/media/72029/Qualifying-for-Pediatric-Exclusivity-Under-Section-505A-of-the-Federal-Food--Drug--and-Cosmetic-Act.pdf
[9] Best Pharmaceuticals for Children Act and Pediatric Research Equity Act _ FDA[EB/OL]. https://www.fda.gov/science-research/pediatrics/best-pharmaceuticals-children-act-and-pediatric-research-equity-act.
[10] Federal Court Invalidates FDA Pediatric Rule, Health Law & Policy Institute[EB/OL]. https://www.law.uh.edu/healthlaw/perspectives/Children/021223Federal.html.
[11] 郑晓琼. 国外儿童用药监管现状[J]. 中国药物经济学, 2011, 28(1): 51-55.
[12] State of Paediatric Medicines in the EU.10 years of the EU Paediatric Regulation[EB/OL]. https://health.ec.europa.eu/system/files/2017-11/2017_childrensmedicines_report_en_0.pdf.
[13] template-scientific-document-part-b-f_en.doc[EB/OL]. https://view.officeapps.live.com/op/view.aspx?src=https%3A%2F%2Fwww.ema.europa.eu%2Fen%2Fdocuments%2Ftemplate-form%2Ftemplate-scientific-document-part-b-f_en.doc&wdOrigin=BROWSELINKEMA.
[14] Pediatric Study Plans_ Content of and Process for Submitting Initial Pediatric Study Plans and Amended Initial Pediatric Study Plans _ FDA[EB/OL]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pediatric-study-plans-content-and-process-submitting-initial-pediatric-study-plans-and-amended.
[15] FDA / EMA Common Commentary on Submitting an initial Pediatric Study Plan (iPSP) and Paediatric Investigation Plan (PIP) for the Prevention and Treatment of COVID-19 [EB/OL]. https:// www.fda.gov/media/138489/download.
[16] Guideline on the format and content of applications for agreement or modification of a paediatric investigation plan and requests for waivers or deferrals and concerning the operation of the compliance check and on criteria for assessing significant studies [EB/OL]. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52014XC0927(01)CELEX_5201.
[17] PMHS Standard Operating Procedure (SOP) for Review of Pediatric Study Plans (PSPs) and Written Requests by the Pediatric Review Committee (PeRC) [EB/OL]. https://www.fda.gov/media/86061/download
[18] PIP assessment procedure[EB/OL]. https://www.ema.europa.eu/en/documents/presentation/presentation-paediatric-investigation-plan-assessment-procedure_en.pdf.
[19] 药监政策速览(第42期)破解儿童用药短缺难题[EB/OL]. https://www.cde.org.cn/main/news/viewInfoCommon/6e103cc9209c2c999e1b458a71bb0eb0.
[20] 2021年度药品审评报告[EB/OL]. https://www.cde.org.cn/main/news/viewInfoCommon/f92b7bdf775bbf4c4dc3a762f343cdc8.
[21] International Collaboration _ Pediatric Cluster _ FDA[EB/OL]. https://www.fda.gov/science-research/pediatrics/international-collaboration-pediatric-cluster.
[22] 国家药监局连任ICH管委会成员 我国药品监管加速国际化进阶[EB/OL]. https://www.cde.org.cn/main/news/viewInfoCommon/a6f9b5e97e82fb01fe205536d06b2c3f.