您当前的位置:检测资讯 > 生产品管
嘉峪检测网 2024-09-29 19:23
持续工艺确认是药品生命周期中商业化生产阶段一项很重要的工作,做好持续工艺确认工作,能够确保药品质量持续稳定,保障人民用药安全。本文通过对国内制药企业在进行药品持续工艺确认过程中存在的主要问题进行研究和分析,并针对问题提出解决的方法和建议,以期为国内制药企业进行药品持续工艺确认工作提供有益参考。
持续工艺确认是对药品上市后的商业化生产直至药品退市全过程开展的验证工作,是药品生命周期工艺验证中最长的阶段。2015 年 12 月 1 日,原国家食品药品监督管理总局发布实施的《确认与验证》附录,在工艺验证章节引入了“持续工艺确认”的概念,要求应当进行持续工艺确认,对商业化生产的产品质量进行监控和趋势分析,以确保工艺和产品质量始终处于受控状态”[1]。该附录仅提出制药企业应进行持续工艺确认的要求,但是未规定具体的实施方法及策略。通过对国内制药企业执行持续工艺确认的调查,发现我国制药企业在执行持续工艺确认过程中存在诸多共性问题。本文围绕存在的问题展开相关研究,结合国内和国际上有关工艺验证指南、法规以及相关文献,提出对我国制药企业执行持续工艺确认改进的思路。
1、我国制药企业执行持续工艺确认的现状
在《药品生产质量管理规范》的确认与验证附录实施前,我国制药企业普遍采用定期工艺再验证或回顾性验证的方式对生产工艺进行确认。而目前无论是国内还是国际有关工艺验证的法规或指南,都明确提出了在药品生命周期中进行“持续工艺确认”的要求,并在 2015 年 10 月 1 日,新修订的欧盟 GMP 附录 15《确认与验证》中已经明确说明不再接受“回顾性验证”的方法,工艺验证应采取持续工艺确认的方法进行[2]。在近几年的 GMP 认证检查过程中,有些制药企业由于未进行持续工艺确认或者持续工艺确认不完善而被列入检查缺陷报告。目前,由于我国缺少指导企业进行持续工艺确认的指南性文件,对于大多数制药企业而言如何有效开展持续工艺确认依然存在困惑。
1.1我国制药企业持续工艺确认存在的问题
2019 年 7 月至 2022 年 7 月,对国内 60 家大中型制药企业的持续工艺确认执行情况进行了调查,其中包括 38 家原料药企业和 22 家制剂企业,通过调查发现,我国制药企业在进行持续工艺确认过程中存在较多问题,见表1。

表1 我国制药企业执行持续工艺确认存在问题调查
1.2我国制药企业执行持续工艺确认的现状分析
对制药企业执行持续工艺确认情况的调查可见,在执行药品持续工艺确认过程中,一部分制药企业的持续工艺确认工作尚未开展,有些制药企业仅仅是为了满足法规的要求,开展了该项工作。我国制药企业在实施持续工艺确认过程中,在数据收集体系的建立、取样及监测计划的制定、数据的判异原则、质量数据分析控制策略、数据回顾分析与报告的周期方面的问题较为突出(分别占 78%,82%,85%,72%和 68%)。
2、存在问题分析及改进建议
持续工艺确认是对生产过程中的数据进行实时收集,并通过统计分析工具判断数据的趋势,及时发现生产过程中的异常并采取纠正预防措施,确保生产持续处于验证状态[3]。在该阶段应该动态监测和持续收集所有生产批次产品的数据,使用合理有效的统计分析工具判断生产过程是否存在偏离或不良趋势,并评估对产品质量属性的影响程度进而采取控制措施,最终达到持续进行工艺改进的目的[4]。持续工艺确认应定期形成报告,下面对制药企业在持续工艺确认过程中存在的问题逐一展开研究并提出改进建议。
2.1建立数据收集体系
2. 1. 1 问题分析
部分药企业面对生产过程中输出的大量数据(包括进厂原辅料、中间体和产品检测指标及工艺控制参数等),不知如何选择持续工艺确认重点监测和分析的数据。产生该问题的原因主要是由于缺乏对产品及生产工艺的理解。
2. 1. 2 改进建议
企业应基于在药品研发和首次工艺验证阶段所获取的知识,选择对产品质量有重大影响的关键数据进行重点监测和统计分析,美国食品和药物管理局(FDA) 推荐使用“质量度量”的方法对持续工艺确认中的数据进行统计和分析[5],企业建立尽可能多的质量度量指标,对产品质量状况进行监测和评价,数据包括产品生产过程中产生的指标类数据及控制药品生产工艺的参数类数据,指标类数据代表药品的安全性和有效性,参数类数据的异常波动会引起药品安全性和/或有效性的问题。在持续工艺确认过程中需重点监测的数据,应至少包括但不限于: (1) 原辅料、中间体、中间产品的关键物料属性(CMAs) 检测数据; (2) 药品的关键质量属性(CQAs) 检测数据; (3)生产过程关键工艺参数(CPPs) 监测数据。
2.2取样及监测计划
2. 2. 1 问题分析
我国大多数制药企业在制定持续工艺确认的取样及监测计划时,未根据风险评估确定监测的范围和频率,也未按照《药品生产质量管理规范》附录: 确认与验证中的要求对监测的范围和频率进行周期性的审核和调整,问题产生的原因一方面是由于缺乏对生产过程的风险控制意识,另一方面是对持续工艺确认中数据监测的目的理解不够深入。
2. 2. 2 改进建议
持续工艺确认监测范围应基于产品知识及对生产工艺的理解,根据风险程度来制定。在产品持续工艺确认过程中,监测的频率应根据对工艺的理解和工艺性能控制水平的变化,进行周期性的审核和调整[1],对于无生产历史的新上市药品,在进入持续工艺确认的初始阶段,需要维持较高的取样及监测频率,直到有足够的统计学数据证明生产过程管控能力持续稳定后,方可降低取样频率; 当发现生产过程出现异常趋势时,应该提高取样及监测的频率,提高异常偏离的可检测性,及时找到可能导致偏离的根源。如某小容量注射剂生产过程中对配液后“灌装装量”的监测,在持续工艺确认时常规监测频率为每小时取样,当监测到“灌装装量”的数据出现异常波动时,及时将取样频率由每小时取样调整为每半小时取样,提高对生产过程的监控,发现问题及时查找原因并采取措施,及时对装量进行控制,待工艺持续稳定后再恢复到常规的取样测试频率。
制药企业应基于风险评估的结果确定取样或测试的手段,如对生产过程中关键工序采用 PAT 分析技术进行实时采集、分析和控制,达到及时分析、控制和稳定生产的目的,如某小容量注射剂产品需要充氮保护,在生产灌装工序后安装在线残氧量的检测装置,实时进行残氧量的检测,及时发现残氧不合格的产品并加以剔除。另外,对于生产过程中某些关键工艺参数的控制,采用对参数进行自动化控制,以确保生产工艺的稳定。
2.3监测数据的判异原则
2. 3. 1 问题分析
制药企业在进行监测数据分析时,出现对监测数据判异原则制定不够合理的现象,将判异原则直接定为不符合产品内控标准,不能及时发现生产过程中的异常趋势或偏离,采取纠偏措施,防止产品不合格的发生。产生该问题的原因一方面是由于人员未结合生产工艺、产品实际质量水平等制定合理的数据判异原则,另一方面,是由于不善于利用合理的统计分析工具进行生产过程的监控及评价,及时识别异常的出现。
2. 3. 2 改进建议
采用何种判异原则应根据生产工艺监测的目的来确定,异常数据包括超趋势结果(Out-of-Trend) 、超标结果(Out-of-Specification) 、超出行动限结果(Out-of-Action Limit) 等[6],对于有具体数值的药品关键质量属性(CQAs) ,如含量等,可使用趋势图、控制图、过程能力分析等统计分析方法[7]。如果使用过程控制图进行生产过程控制,控制图有8种判异原则,可根据对产品工艺的理解并结合生产控制需求选择其中的一种或几种判异原则进行控制。
2.4建立质量数据分析控制策略
2. 4. 1 问题分析
制药企业在药品持续工艺确认过程中出现数据可靠性的问题,考虑主要集中在编造或篡改数据、原始数据无法溯源、电子数据安全性不足等方面。另外,企业在持续工艺确认过程中统计分析工具使用不当,会导致不能及时发现生产过程中的异常数据,对异常情况不能及时进行分析和调查并制定有效的纠正和预防措施,最终会导致产品质量发生异常,给人民用药的安全性和有效性造成风险,也会给企业带来巨大损失。
2. 4. 2 改进建议
生产过程中采集到的数据可以采用人工记录或电子记录的方式,无论采用何种方式对数据进行采集和记录,都必须确保数据的可靠性,保证数据符合美国 FDA 所提出的 ALCOA 原则,即数据要遵循可追溯、易读性、即时性、原始性和准确性,只有这样的数据,才能确保数据具有统计和分析的意义,数据分析应具备以下前提[8]: (1) 数据可靠性有保证,检测、监测方法可靠,分析方法经过验证,用于监视测量的仪器、仪表均经过校准,参与记录的人员均经过数据可靠性的培训,确保所获得的结果是有分析价值的数据; (2) 进行统计学分析的指标对于期望获得的信息是有意义的; (3) 应用统计工具的投入程度、工具选择以及记录应与风险的级别相适应。
制药企业应使用合理有效的统计分析工具对生产过程中收集到的数据进行分析,FDA 推荐企业使用定量的统计学方法,以便于能及时发现生产过程中出现的不良趋势,有效评价生产过程是否持续处于验证状态,持续工艺确认阶段可以运用的统计分析工具有[9]: (1) 运用工序能力指数(Cpk) 分析工艺控制能力是否足够; (2) 运用统计控制图(SPC) 进行过程控制分析; (3) 出现变更时,运用方差分析、t 检验等统计分析工具,将变更前后产品质量情况进行对比分析; (4) 运用回归分析的方法对产品稳定性数据进行分析,预测产品在有效期内是否合格。
以某制剂产品“含量”监测为例,使用控制图联合 Minitab 分析软件对生产过程进行控制,对于生产过程的失控状态实时报警,及时进行原因分析并采取纠偏措施[10],下图为对连续 50 个批次的产品“含量”指标进行连续监测,使用 Minitab 软件制作图1 中的含量分析六合图[11]。
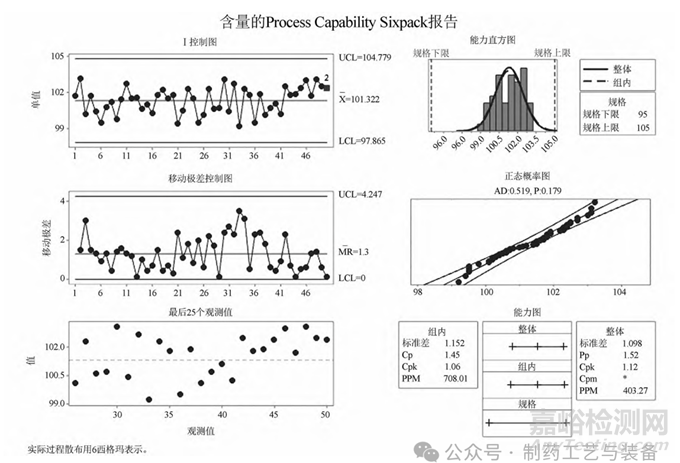
图1 含量分析六合图
以上六合图包含了单值-移动极差的 I-MR 控制图、能力直方图、正态概率图、能力图和最后 25 个观测值的运行图,分析如下:
(1) 控制图分析:设定控制图的判异原则为单点超出上控制限(UCL) 或下控制限(LCL) 和连续 9 个点位于中心线的同一侧,选择这两种判异原则的依据是单点超标即判异,连续 9 个点位于中心线的同一侧意味着“含量”从随机波动的状态进入异常波动,意味着过程出现异常,需要查找原因,必要时采取纠正预防措施。从 I-MR 控制图可以看到,过程中有连续9 个点位于中心限同一侧的情况(出现红色报警信号2) ,故判为异常,经调查该 9 个批次的产品,为含量检验方法由原来的生物效价法改为了 HPLC 法所致,在以后的生产中应持续观察,根据趋势情况,必要时对产品内控标准进行修订。
(2) 从能力直方图、正态概率图和能力图可以看出,该产品 50 个批次的“含量”数据 P = 0. 179 > 0. 05,数据呈现正态分布。
(3) Cp 为表示过程加工一致性的指数,即表示“质 量 能 力 ”,从图中可以看出 Cp = 1. 45,为1. 33≤Cp < 1. 67,过程能力充分,另外,得出 Cpk =1. 06,按照表2将 Cp 与 Cpk 联合使用后,得出合格品率 大 约 为 99. 87% (表2中斜体数值),该数值 > 99. 73% ,符合 3σ 控制原则,说明产品生产过程较为稳定。
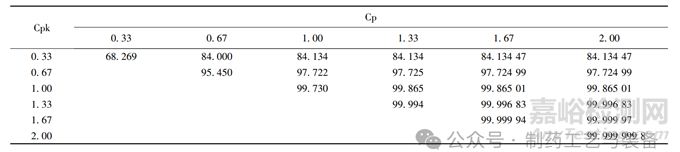
表2 联合应用Cp与Cpk所代表的合格品率[12](%)
2.5持续工艺确认数据回顾分析与报告
2. 5. 1 问题分析
我国很多制药企业对持续工艺确认数据的报告频率制定不合理,有些企业采取年度回顾的方式进行,不能保证及时发现生产过程中的变化趋势,并提出相应的改进措施。
2. 5. 2 改进建议
应该对持续工艺确认过程中收集到的数据进行定期的回顾分析并形成报告,回顾和报告的目的是对一定生产周期内工艺运行情况和所生产的产品进行分析和评价,并提出对下一阶段持续改进的建议。回顾的频率应基于风险及生产过程控制的复杂程度和可控程度,对于有很长的生产历史且质量稳定的产品来说,回顾和报告的频率可以稍低,而对于新产品,历史数据少,对产品和工艺的理解不够,应该以更高频率进行回顾和报告,以便于及时发现工艺变化的趋势,待积累较多的生产数据,对产品及工艺的理解逐渐深入后,可降低产品持续工艺确认回顾分析和报告的频率。
3、对执行持续工艺确认的建议
以上内容对我国制药企业药品持续工艺确认的现状及存在的问题进行了分析,针对所存在的问题,结合国内外有关药品工艺验证的法规、指南及相关文献等,开展了药品持续工艺确认的系统研究工作,对如何做好药品持续工艺确认工作提出以下几点思考及改进措施:
3.1对我国制药企业的建议
3. 1. 1 建立基于风险管理的工艺验证体系
制药企业应建立基于风险管理的工艺验证体系,根据对产品和工艺的理解,将质量风险管理融入到药品持续工艺确认过程中。在持续工艺确认过程中,制药企业应重视统计分析工具的应用,通过采用先进的生产技术和分析技术,如“过程分析技术(PAT)”,持续不断地获取质量和工艺数据,进行统计学趋势分析,及时预知异常趋势,采取有效的控制措施,预防由于潜在的异常事件积累而导致对产品质量的 影 响,确保药品生产工艺稳定和持续改进。
3. 1. 2 加强知识管理
制药企业应加强药品生命周期的知识管理,在产品自研发阶段向商业化生产阶段转移时,做好技术衔接,确保在产品研发阶段所积累的产品及工艺知识有效传递至产品商业化生产阶段[13]。在上市后的商业化生产阶段,应通过持续工艺确认对生产工艺进行持续监测,发现异常并对生产工艺不断进行改进,制定相应的控制策略,并将相关知识补充到产品档案中,不断加强对产品知识的管理。
3. 1. 3 严格变更管控
在进行持续工艺确认过程中,由产品质量改进、工艺创新等均可引发变更,在变更实施过程中可能会给药品质量带来潜在风险。企业需构建以质量风险管控为核心的变更管控系统[14]。目前我国已发布很多药品上市后变更法规和指导性文件,2017 年,ICH 发布了 Q12(《药品生命周期管理的技术和监管考虑》) 草案,该草案有助于创建更便利的批准后变更管理体系,并且强调加强知识管理,保证整个产品生命周期内知识的连续性,并且鼓励企业将 QbD 的理念用于批准后变更管理方案(PACMP) [15]。我国制药企业应严格执行我国对上市后药品变更的管理要求,并参照 ICHQ12 的要求严格进行变更管控。
3.2为药品监管决策提供参考
可结合我国国情和制药企业的实际情况,制定与国际接轨的科学的、系统化的药品持续工艺确认指南性文件,促进我国制药企业提高对药品持续工艺确认的认识和重视程度,逐步推进药品持续工艺确认工作与国际接轨,稳定和提高药品质量。
4、结 语
本研究针对我国制药企业在持续工艺确认过程中存在的问题,结合我国制药企业的现状进行了研究,该研究为我国制药企业在持续工艺确认过程中存在的问题提供了解决方案。但新法规和要求的推行需要我国监管机构和制药企业的共同努力,各项措施的实施也需要一定的时间,对我国大多数制药企业来说,可以借鉴国际工艺验证的法规及指南的要求,成立专家团队,进行内部培训,更加有效地开展药品持续工艺确认工作。
参考文献
[1] 原国家食品药品监督管理总局 . 药品生产质量管理规范(2010 年修订)GMP 附录: 确认与验证[EB/OL]. (2015-5-26](2022-12-1]. https: / /www. nmpa. gov. cn /yaopin /ypggtg /ypqtgg /20150526120001509. html
[2] European Commission. EU GMP: Annex 15: Qualification and Validation[EB/OL]. (2015-03-30) [2015-10-1]. https: / /health. ec. europa. eu /system /files/2016-11 /2015-10_annex15_0. pdf[3] 刘炳坤.《持续工艺确认》的发展及实施要点[J]. 海峡药学,2019,31(9) : 292-294.
[4] 王亚涛,赵九洲,靳杰,等. 新版 GMP 附录中“持续工艺确认”方法探析[J]. 机电信息,2016,(5) : 12-14,58.
[5] USFDA. Submission of Quality Metrics Data Guidance for Industry(DRAFTGUIDANCE) [EB/OL]. [2018-08-29]https: / /www. fda. gov /downloads/drugs/guidancecomplianceregulatoryinformation /guidances/ucm455957. pdf
[6] PDA. Process Validation A Lifecycle Approach[EB/OL]. (2013]( 2017-10-18]. http: / /www. pda. org /pcmo
[7] 孙宾,王亚锴,郭荣志,等. 持续工艺确认执行策略及实施应用[J]. 流程工业,2020(2) : 23-27.[8] PDA. Technical Report No. 59 Utilization of Statistical Methods For Production Monitoring [EB/OL]. (2018-06-25) [2022-10-26]. http: / /www.pda. org /pcmo
[9] 肖志坚,药品生产质量控制统计学方法及案例分析[J]. 第九届江苏省药师周大会论文集,2009: 360-365
[10] 王春涛,唐静,陈伟. Minitab 软件在药品生产质量控制中的应用[J]. 中国执业药师,2012,107(11) : 42-46.[11] 朱存节,林娟. Minitab 软件在制药企业产品质量回顾分析中的应用[J]. 中国药物经济学,2013(S3) : 19-21.
[12] 中级/全国质量专业技术人员执业资格考试办公室. 质量专业理论与实务[M]. 中国人事出版社,2007: 182-185
[13] 丁恩峰,高海燕 EMA 最新工艺验证指南解读[J]. 医 药 工 程 设 计,2012,33(5) : 37-41.
[14] 李晓宇,柴倩雯,田德龙,等. 欧美已批药品生产变更研究[J]. 中国药物警戒,2016,13(8) : 476-481.
[15] ICH harmonized tripartite guideline. Technical and Regulatory Considerationsfor Pharmaceutical ProductLifecycle Management,Q12[EB/OL]. (2019)[2019-11-20]. https: / /database. ich. org /sites/default /files/Q12 _ Guideline_Step4_2019_1119. pdf
本文作者贾欣秒、赵建强、李红秀,华北制药股份有限公司、华北制药华胜有限公司,来源于中国药物评价,仅供交流学习。

来源:Internet


