您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-10-30 16:05
国家药监局组织起草了《化妆品中壬二酸及其盐类的检验方法》等5项检验方法,经国家药监局化妆品标准化技术委员会主任会议审查通过,现予以发布。
《化妆品中壬二酸及其盐类的检验方法》《化妆品中非那西丁的检验方法》《化妆品中羟基癸酸的检验方法》等3项检验方法为新增的化妆品检验方法,纳入《化妆品安全技术规范(2015年版)》,自2025年7月1日起施行。
《化妆品中石棉的检验方法》《化妆品中葡糖醛酸等14种原料的检验方法》为修订的化妆品检验方法,替换《化妆品安全技术规范(2015年版)》中原有检验方法;自2025年7月1日起,化妆品注册、备案及抽样检验等相关检验应当采用本通告发布的上述两个检验方法。
《化妆品安全技术规范》5项标准制修订项目汇总表
|
序号 |
项目名称 |
类型 |
建议纳入《化妆品安全技术规范》的章节 |
同时废止的《化妆品安全技术规范》中原章节内容 |
|---|---|---|---|---|
|
1 |
化妆品中壬二酸及其盐类的检验方法 |
新增检验方法 |
第四章理化检验方法8 其他原料检验方法8.4 化妆品中壬二酸及其盐类的检验方法 |
|
|
2 |
化妆品中非那西丁的检验方法 |
第四章理化检验方法2 禁用组分检验方法2.39 化妆品中非那西丁的检验方法 |
|
|
|
3 |
化妆品中羟基癸酸的检验方法 |
第四章理化检验方法3 限用组分检验方法3.14 化妆品中羟基癸酸的检验方法 |
|
|
|
4 |
化妆品中石棉的检验方法 |
修订后替换原检验方法 |
第四章理化检验方法2 禁用组分检验方法2.27 化妆品中石棉的检验方法 |
第四章理化检验方法2 禁用组分检验方法2.27 石棉 |
|
5 |
化妆品中葡糖醛酸等14种原料的检验方法 |
第四章理化检验方法3 限用组分检验方法3.1 化妆品中葡糖醛酸等14种原料的检验方法 |
第四章理化检验方法3 限用组分检验方法3.1 α-羟基酸(国家药品监督管理局2019年第12号通告) |
化妆品中壬二酸及其盐类的检验方法
Determination of Azelaic Acid and its salts in Cosmetics
1 范围
本方法规定了高效液相色谱法测定化妆品中壬二酸及其盐类(以壬二酸计)的含量。
本方法适用于膏霜乳类、液体类、凝胶类、粉剂类、泥类、贴膜类化妆品中壬二酸及其盐类(以壬二酸计)的测定。
2 方法提要
样品提取净化后,经高效液相色谱仪分离,二极管阵列检测器检测,根据保留时间和紫外光谱定性,峰面积定量,以标准曲线法计算含量。
本方法壬二酸的检出限为0.05 μg,定量下限为0.17 μg;当取样量为1.0 g,稀释定容体积为20 mL时壬二酸的检出浓度为0.005%,最低定量浓度为0.017%。
3 试剂和材料
除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T 6682规定的一级水。
3.1 甲醇。
3.2 氨水。
3.3 磷酸。
3.4 乙腈,色谱纯。
3.5 氨溶液:量取氨水(3.2)1 mL,用水稀释至100 mL。
3.6 2%磷酸甲醇溶液:量取磷酸(3.3)2 mL,用甲醇(3.1)稀释至100 mL。
3.7 0.1%磷酸溶液:量取磷酸(3.3)1.0 mL,用水稀释至1000 mL。
3.8 标准品:壬二酸(英文名称:Azelaic Acid,分子式:C9H16O4;分子量:188.22;CAS号:123-99-9)。
3.9 壬二酸标准储备溶液:称取壬二酸标准品40 mg(精确到0.00001 g)于10 mL容量瓶中,用水溶解并稀释至刻度[可先加少量甲醇(3.1)超声助溶],摇匀,即得浓度为4 mg/mL的壬二酸标准储备溶液,避光密封保存,20日内稳定。
4 仪器和设备
4.1 高效液相色谱仪,二极管阵列检测器。
4.2 天平,感量0.0001 g和0.00001 g。
4.3 超声波清洗器,功率≥120 W。
4.4 涡旋振荡器。
4.5 离心机。
4.6 混合阴离子固相萃取柱(填料质量≥150 mg),基质为季铵盐改性的聚苯乙烯-二乙烯基苯高聚物,使用前依次用15 mL甲醇(3.1)、15 mL水活化。
4.7 固相萃取装置。
5 分析步骤
5.1 标准系列溶液的制备
分别准确移取壬二酸标准储备溶液(3.9)适当量,用水稀释配制成浓度分别为3 μg/mL、10 μg/mL、20 μg/mL、50 μg/mL、100 μg/mL的标准系列溶液。
5.2 样品处理
5.2.1 洗面奶、洗发水、沐浴露等淋洗类化妆品
称取混合均匀的样品1.0 g(精确到0.0001 g)于10 mL具塞比色管中,加甲醇(3.1)约8 mL,涡旋分散后超声提取10 min,放冷后定容至刻度,准确量取5 mL上清液(必要时可离心或过滤),置10 mL具塞比色管中,用水定容至刻度,摇匀,滤过,即得待测溶液。
5.2.2 其他类化妆品
称取混合均匀的样品1.0 g(带有载体的面膜,去除载体后取样;精确到0.0001 g)于10 mL具塞比色管中,加甲醇(3.1)2 mL,涡旋分散,加水约6 mL,加氨溶液(3.5)0.5 mL,充分涡旋2 min后超声提取10 min,放冷后用水定容至刻度,涡旋混匀。准确量取0.5~5 mL上清液(必要时可离心或过滤)置已活化好的固相萃取柱(4.6)上,待样品溶液流尽后,依次用水15 mL、甲醇(3.1)15 mL淋洗固相萃取柱,弃去淋洗液,再用2%磷酸甲醇溶液(3.6)5 mL洗脱,收集洗脱液置10 mL比色管中,用水定容至刻度,摇匀,滤过,即得待测溶液。
注1:待测原料含量比较大的样品可通过适当稀释或减少上样体积以确保待测溶液浓度在标准曲线范围内。
注2:凝胶类样品推荐上样量不超过2 mL。
5.3 参考色谱条件
色谱柱:金刚烷基键合硅胶色谱柱(5 μm,4.6×250 mm),或等效色谱柱;
流动相:A:乙腈(3.4);B:0.1%磷酸溶液(3.7);梯度洗脱程序见表1。
表1 梯度洗脱程序
|
时间/min |
V(A)/% |
V(B)/% |
|---|---|---|
|
0 |
17 |
83 |
|
6 |
17 |
83 |
|
8 |
26 |
74 |
|
15 |
26 |
74 |
|
17 |
90 |
10 |
流速:1.0 mL/min;
柱温:40 ℃;
进样量:20 μL;
检测波长:210 nm。
5.4 测定
在“5.3”色谱条件下,取标准系列溶液(5.1)分别进样,进行色谱分析,以标准系列溶液浓度为横坐标,峰面积为纵坐标,绘制标准曲线。
取“5.2”项下的待测溶液进样,根据保留时间和紫外光谱图定性,测得峰面积,根据标准曲线得到待测溶液中壬二酸的浓度。按“6”计算样品中壬二酸的含量。
6 分析结果的表述
6.1 计算
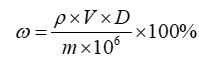
式中:ω(以壬二酸计)——化妆品中壬二酸的质量分数,%;
ρ——从标准曲线得到壬二酸的质量浓度,μg/mL;
V——样品稀释定容体积,mL;
m——样品取样量,g;
D——稀释倍数(不稀释则取1)。
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
6.2 回收率和精密度
多家实验室的回收率在80.7%~110.6%之间,相对标准偏差小于9.2%(n=6)。
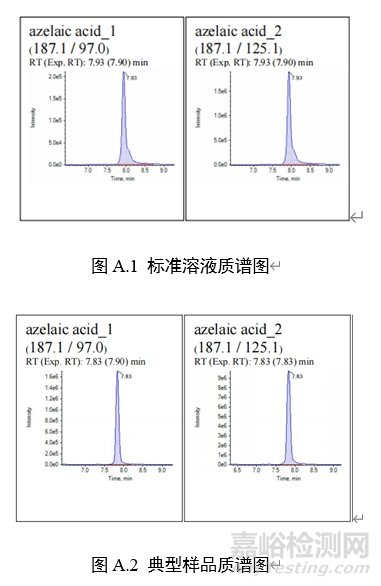
7 图谱

1:壬二酸(15.8min)
注:壬二酸二钾和壬二酸二钠在液相色谱、液相色谱-质谱上均是以壬二酸形式被检出。
附录A
(规范性附录)
壬二酸结果的确证
如液相色谱方法中检出结果和化妆品标签成分不一致,需采用液相色谱-质谱法进行确认。
A.1 样品前处理过程见5.2样品处理。
A.2 参考色谱条件
色谱柱:C18柱(1.8 μm,2.1×100 mm),或等效色谱柱;
流动相:A:乙腈;B:0.05%甲酸溶液。
梯度洗脱程序见表A.1;
表A.1 流动相梯度洗脱程序
|
时间/min |
V(A)/% |
V(B)/% |
|
0 |
35 |
65 |
|
3 |
35 |
65 |
|
7 |
90 |
10 |
|
8 |
90 |
10 |
|
8.1 |
35 |
65 |
|
10.5 |
35 |
65 |
流速:0.3 mL/min;
柱温:40 ℃;
进样量:2 μL。
A.3 参考质谱条件
离子源:电喷雾离子源(ESI);
监测模式:负离子多反应监测模式(MRM);监测离子对及相关电压参数设定见表A.2;
表A.2 监测离子对及相关电压参数设定表
|
名称 |
母离子(m/z) |
子离子(m/z) |
碰撞能(V) |
|
壬二酸 |
187.1 |
125.1* |
-20 |
|---|---|---|---|
|
97.0 |
-24 |
*为推荐定量离子。
注:当采用不同质谱仪器时,仪器参数可能存在差异,测定前应将质谱参数优化到最佳。
A.4 定性
用液相色谱-三重四极杆串联质谱仪进行定性。如样品检出的特征离子峰的保留时间与浓度相近的壬二酸标准溶液一致(相对偏差在±2.5%之内),且样品所选择的监测离子对的相对丰度比与标准溶液的监测离子对的相对丰度比的偏差不超过表A.3规定范围,则可判断样品中存在壬二酸。
表A.3 结果确证时离子对相对丰度比的最大允许偏差
|
相对离子丰度(k) |
k>50% |
50%≥k>20% |
20%≥k>10% |
k≤10% |
|
允许的最大偏差 |
±20% |
±25% |
±30% |
±50% |
化妆品中非那西丁的检验方法
Determination of Phenacetin in Cosmetics
1 范围
本方法规定了液相色谱-串联质谱法测定化妆品中非那西丁的含量。
本方法适用于膏霜乳类、液体类、凝胶类、粉类等化妆品中非那西丁的定性与定量。
2 方法提要
样品以乙腈为溶剂提取,采用高效液相色谱仪分离,质谱检测器检测,根据保留时间和特征离子对的相对丰度比定性、定量离子对峰面积定量,以标准曲线法计算含量。
本方法取样量为0.5 g时,检出浓度为0.012 mg/kg,最低定量浓度为0.04 mg/kg。
3 试剂和材料
除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T 6682规定的一级水。
3.1 甲醇,色谱纯。
3.2 乙腈,色谱纯。
3.3 氯化钠。
3.4 50%乙腈:取乙腈、水等体积混合,摇匀。
3.5 饱和氯化钠溶液:称取40 g氯化钠(3.3),置于250 mL磨口锥形瓶中,加入100 mL水,超声15 min,即得。
3.6 标准品:非那西丁(英文名称:Phenacetin,分子式:C10H13NO2,CAS:62-44-2),纯度≥98%。
3.7 标准储备溶液:精密称取非那西丁标准品10 mg(精确到0.00001 g)于10 mL容量瓶中,加入甲醇(3.1)溶解并定容,配制成质量浓度为1 mg/mL的标准溶液备用,转入棕色储液瓶中,4 ℃避光保存,有效期3个月。
3.8 标准中间液:精密量取适量标准储备溶液(3.7),用甲醇(3.1)稀释成质量浓度为10 mg/L的标准中间液。
4 仪器和设备
4.1 高效液相色谱-三重四极杆质谱联用仪。
4.2 分析天平:感量0.0001 g和0.00001 g。
4.3 超声波清洗器(≥250 W)。
4.4 涡旋混合仪。
4.5 高速离心机。
5 分析步骤
5.1 空白基质提取液
称取空白试样0.5 g(精确到0.0001 g),置于15 mL具塞比色管中,自“加入饱和氯化钠溶液2 mL”起与样品同法处理(5.4),作为空白基质提取液。该基质提取液进入质谱后,在非那西丁的特征离子峰保留时间处未出峰且未检出其特征离子对,则确认为空白基质提取液,可用于下一步实验。空白样品的性状应与待测化妆品尽量基本一致。
5.2 基质标准中间液
精密量取非那西丁标准中间液(3.8)0.1 mL,置于10 mL容量瓶中,用空白基质提取液(5.1)稀释至刻度,摇匀,制成非那西丁浓度为100 μg/L的基质标准中间液。基质标准中间液现用现配。
5.3 基质标准系列溶液的制备
精密量取基质标准中间液(5.2)0.1、0.2、0.5、1、2、5 mL,用空白基质提取液(5.1)配制浓度为1.0、2.0、5.0、10、20、50 μg/L基质标准系列溶液。基质标准系列溶液现用现配。
5.4 样品处理
膏霜乳类、液体类、凝胶类:称取样品0.5 g(精确到0.0001 g),置于15 mL离心管中,加入饱和氯化钠溶液(3.5)2 mL,涡旋30 s,分散均匀,准确加入乙腈(3.2)10 mL,涡旋30 s,超声提取10 min,静置至室温,以8000 r/min转速离心5 min。准确吸取上清液5 mL,加50%乙腈(3.4)定容至10 mL,混匀,经0.22 μm滤膜过滤后,滤液作为供试品溶液备用(供试品溶液可根据实际浓度进行适当稀释)。
粉类:称取样品0.5 g(精确到0.0001 g),置于15 mL离心管中,准确加入乙腈(3.2)10 mL,涡旋30 s,超声提取10 min,静置至室温,以8000 r/min转速离心5 min。准确吸取上清液5 mL,加50%乙腈(3.4)定容至10 mL,混匀,经0.22 μm滤膜过滤后,滤液作为供试品溶液备用(供试品溶液可根据实际浓度进行适当稀释)。
5.5 参考液相色谱-三重四极杆质谱联用条件
色谱条件
色谱柱:C18色谱柱(2.1 mm×100 mm,2.7 μm),或等效色谱柱;
柱温:30 ℃;
进样体积:2 μL;
流动相A:水,流动相B:乙腈;
流速:0.3 mL/min。
表1 梯度洗脱程序
|
时间(min) |
流动相A(%) |
流动相B(%) |
|---|---|---|
|
0.0 |
90 |
10 |
|
2.0 |
90 |
10 |
|
5.0 |
25 |
75 |
|
5.1 |
10 |
90 |
|
7.0 |
10 |
90 |
|
7.1 |
90 |
10 |
|
9.0 |
90 |
10 |
质谱条件
离子源:电喷雾离子源(ESI源);
监测模式:正离子多反应监测模式(MRM),监测离子对及相关参数设定见表2。
表2 非那西丁的监测离子对及相关参数设定
|
组分名称 |
母离子 (m/z) |
子离子 (m/z) |
CE (V) |
|---|---|---|---|
|
非那西丁 |
180.1 |
110.2* |
26 |
|
138.1 |
14 |
*为推荐的定量离子。
注:当采用不同质谱仪器时,仪器参数可能存在差异,测定前应将质谱参数优化到最佳。
5.6 定性判定
取供试品溶液(5.4)与标准溶液在相同分析条件下测定,样品中如呈现定量离子对和定性离子对的色谱峰,非那西丁的特征离子峰保留时间与标准溶液对应的保留时间偏差小于±2.5%,且选择的监测离子对的相对丰度比与相当浓度的标准溶液的监测离子对的相对丰度比的最大偏差不超过表3的规定,则可以判定样品中存在非那西丁。
表3 定性确证时相对离子丰度比的最大允许偏差
|
相对离子丰度(k) |
k>50% |
50%≥k>20% |
20%≥k>10% |
k≤10% |
|
允许的最大偏差 |
±20% |
±25% |
±30% |
±50% |
5.7 定量测定
在“5.5”项液相色谱-三重四极杆质谱联用条件下,取基质标准系列溶液(5.3)依次测定,以非那西丁的系列浓度为横坐标,其定量离子对色谱峰面积为纵坐标,进行线性回归,绘制基质标准曲线,其线性相关系数应大于0.99。
取“5.4”项下处理得到的待测溶液进样,将非那西丁定量离子对色谱峰面积代入基质标准曲线,计算非那西丁的质量分数,按“6”项下公式,计算样品中非那西丁的含量。
6 分析结果的表述
6.1 计算

式中:ω—样品中非那西丁的质量分数,mg/kg;
ρ—供试品溶液中非那西丁的质量浓度,μg/L;
V—样品定容体积,mL;
m—样品取样量,g;
D—稀释倍数(如未稀释则为1)。
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%。
6.2 回收率和精密度
多家实验室验证回收率为82.6%~115.8%,相对标准偏差小于6.4%。
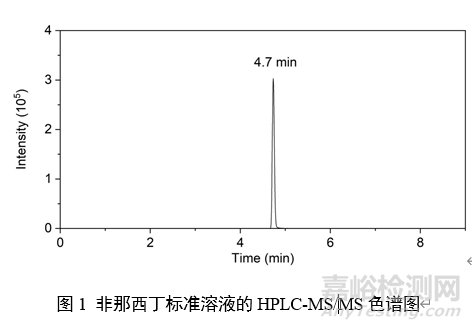
7 图谱
Determination of Hydroxycapric Acid in Cosmetics
1 范围
本方法规定了高效液相色谱法测定化妆品中羟基癸酸的含量。
本方法适用于液体类、凝胶类、膏霜乳类化妆品中羟基癸酸含量的测定。
2 方法提要
以甲醇提取化妆品中羟基癸酸,用高效液相色谱仪进行分离,二极管阵列检测器检测,以保留时间和紫外光谱图定性,峰面积定量,以标准曲线法计算含量。
本方法中羟基癸酸的检出限为0.125 μg、定量下限为0.375 μg。取样量为1.0 g,定容体积为10 mL时检出浓度为250 μg/g,最低定量浓度为750 μg/g。
3 试剂和材料
除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T 6682规定的一级水。
3.1 磷酸氢二铵。
3.2 磷酸,优级纯。
3.3 甲醇,色谱纯。
3.4 乙腈,色谱纯。
3.5 甲酸,色谱纯。
3.6 0.05 mol/L的磷酸氢二铵溶液:称取6.60 g磷酸氢二铵(3.1),加水1000 mL溶解,用0.22 μm滤膜过滤。
3.7 标准品:羟基癸酸(英文名称:Hydroxycapric acid,分子式:C10H20O3,CAS:5393-81-7),纯度≥98%。
3.8 标准储备溶液
精密称取羟基癸酸标准品(3.7)0.5 g(精确到0.0001 g)于10 mL容量瓶中,加甲醇(3.3)溶解并定容至刻度,摇匀,配制成标准储备溶液备用,转入棕色储液瓶中,4 ℃避光保存,有效期1个月。
4 仪器和设备
4.1 高效液相色谱仪,二极管阵列检测器。
4.2 天平,感量分别为0.1 mg、0.01 mg。
4.3 超声波清洗器,功率100 W。
4.4 高速离心机,转速≥14000 r/min。
4.5 pH计,精度0.01。
4.6 涡旋振荡器。
5 分析步骤
5.1 标准系列溶液的制备
精密移取不同体积的标准储备溶液(3.8),用甲醇(3.3)配制成浓度为0.125、0.25、0.5、1、2.5、5 mg/mL的标准系列溶液。
5.2 样品处理
称取混合均匀的样品1.0 g(精确到0.001 g)于10 mL具塞比色管中,加甲醇(3.3)至10 mL刻度,充分涡旋混合30 s,超声提取30 min,取适量样品在10000 r/min下高速离心15 min,取上清液过0.45 μm的滤膜,取续滤液作为待测溶液。
5.3 参考色谱条件
色谱柱:C18柱(250 mm×4.6 mm,5 μm),或等效色谱柱;
流动相:甲醇+ 0.05 mol/L的磷酸氢二铵溶液(3.6)[磷酸(3.2)调pH值为3.0](70+30);
流速:1.0 mL/min;
检测波长:214 nm;
柱温:室温;
进样量:5 μL。
5.4 测定
在“5.3”色谱条件下,取标准系列溶液(5.1)分别进样,进行色谱分析,以标准系列溶液浓度为横坐标,峰面积为纵坐标,绘制标准曲线。
取“5.2”项下的待测溶液进样,根据保留时间和紫外光谱图定性,测得峰面积,根据标准曲线得到待测溶液中羟基癸酸的浓度。按“6”计算样品中羟基癸酸的含量。
6 分析结果的表述
6.1 计算
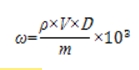
式中:ω——样品中羟基癸酸的质量分数,μg/g;
ρ——从标准曲线得到待测原料的质量浓度,mg/mL;
V——样品定容体积,mL;
m——样品取样量,g;
D——稀释倍数(不稀释则取1)。
在重复性条件下获得的两次独立测试结果的绝对差值不得超过算术平均值的10%。
6.2 回收率和精密度
方法回收率为85%~115%,相对标准偏差小于10%。
7 图谱
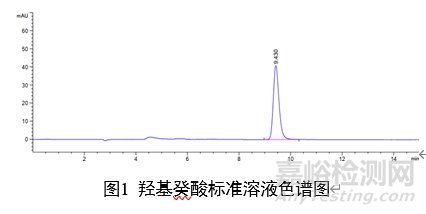
附录A
(规范性附录)
羟基癸酸阳性结果的确证
因羟基癸酸极性大,紫外吸收接近截止波长,测定时易出现干扰,必要时可采用液相色谱-质谱法确证结果,以排除基质中其他组分的干扰。在相同的液相色谱-质谱实验条件下,如果样品中色谱峰的保留时间和紫外光谱图与标准溶液中对应成分一致(相对偏差在±2.5%之内),所选择的监测离子对的相对丰度比与相当浓度标准溶液的离子对的相对丰度比的偏差不超过表A.1规定范围,则可以判定样品中存在对应的测定成分。
表A.1 结果确证时相对离子丰度比的最大允许偏差
|
相对离子丰度(k) |
k>50% |
50%≥k>20% |
20%≥k>10% |
k≤10% |
|
允许的最大偏差 |
±20% |
±25% |
±30% |
±50% |
A.1 仪器参考条件
A.1.1 色谱条件
色谱柱:C18柱(100 mm×2.1 mm,1.8 μm),或等效色谱柱;
流动相:A:乙腈(含0.1%甲酸);B:0.1%甲酸水溶液;
表A.2 流动相梯度洗脱程序
|
时间/min |
V(流动相A)/% |
V(流动相B)/% |
|
0 |
0 |
100 |
|
4 |
0 |
100 |
|
15 |
95 |
5 |
|
17 |
95 |
5 |
|
17.1 |
0 |
100 |
|
22 |
0 |
100 |
流速:0.2 mL/min;
柱温:30 ℃;
进样量:5 μL。
A.1.2 质谱条件
离子源:电喷雾离子源(ESI源);
监测模式:负离子多反应监测模式;监测离子对及相关电压参数设定见表A.3;
鞘气流速:11 L/min;
干燥气流速:14 L/min;
鞘气温度:350℃;
干燥气温度:230℃;
碰撞气:Ar,20 psi;
离子源电压(IS)电压:负离子:3000 V。
表A.3 羟基癸酸的监测离子对及相关电压参数设定表
|
组分名称 |
母离子(m/z) |
子离子(m/z) |
CE(V) |
|
羟基癸酸 |
187.1 |
141.1* |
-20 |
|
139 |
-20 |
*为推荐的定量离子。
注:当采用不同质谱仪器时,仪器参数可能存在差异,测定前应将质谱参数优化。
化妆品中石棉的检验方法
Determination of Asbestos in Cosmetics
1 范围
本方法规定了用偏光显微镜、扫描电子显微镜-能谱仪和X射线衍射仪测定化妆品中石棉的方法。
本方法适用于粉剂类、块状类、膏霜乳类化妆品中石棉的测定。
2 方法提要
石棉具有耐高温性,并具有特定的形态特征、光学特性和元素组成。样品经高温处理和酸洗,使用偏光显微镜检测其中是否含有符合石棉形态特征及光学特性的纤维颗粒。使用扫描电子显微镜-能谱仪对纤维颗粒的形态特征和元素组成进行确认,判定样品中是否含有某种类型的石棉。
石棉X射线衍射峰的强度与其含量成正比。当观测到可明确归属为某一类石棉的X射线衍射峰时,可基于衍射峰的强度确定样品中石棉的含量。
3 术语和定义
石棉是指具有纤维状晶体生长习性的蛇纹石和角闪石类的硅酸盐矿物,包括纤维状蛇纹石(温石棉)和纤维状角闪石类(青石棉、铁石棉、直闪石石棉、透闪石石棉、阳起石石棉)。
4 试剂和材料
除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T 6682规定的一级水。
4.1 石棉标准物质/标准样品:温石棉、青石棉、铁石棉、直闪石石棉、透闪石石棉、阳起石石棉。
4.2 折射率浸油:nD25℃=1.550、1.620、1.690,精度为±0.005。
4.3 甲酸。
4.4 20%甲酸溶液:吸取甲酸(4.3)20 mL,加水至100 mL,摇匀。
4.5 刚玉(α-Al2O3):纯度≥99.9%,粒径≤0.040 mm。
4.6 载玻片和盖玻片:无色透明玻璃,表面洁净,无可见的雾状物、水迹和指印等,无可见的凹坑、粒状物、结石、划痕、断裂等缺陷。
4.7 工具:坩埚、玛瑙研钵、标准筛(325目,孔径45 μm)、移液器、导电胶带、蒸发皿等。
5 仪器和设备
5.1 偏光显微镜(PLM):应达到的技术要求见附录B.1。
5.2 扫描电子显微镜-能谱仪(SEM-EDS):应达到的技术要求见附录C.1。
5.3 X射线衍射仪(XRD):应达到的技术要求见附录D.1。
5.4 分析天平:感量0.1 mg。
5.5 箱式高温炉:温度范围100~500 ℃,控温精度±10 ℃,具备程序升温功能。
5.6 恒温烘箱:温度范围50~120 ℃,控温精度±2 ℃。
5.7 超声波清洗器,功率500 W。
5.8 涡旋混合器。
6 分析步骤
6.1 样品处理
6.1.1 向洁净的坩埚(4.7)中称取约10 g化妆品样品,置于箱式高温炉中。主成分为滑石粉的爽身粉样品可直接升温至465 ℃保持4 h。含有机成分较多的样品可参考表1进行程序升温,必要时应增大称样量,确保灰化后残留物不少于200 mg。
表1 程序升温表
|
步骤 |
起始温度(℃) |
升温时间(min) |
目标温度(℃) |
保持时间(min) |
|
1 |
室温 |
20 |
120 |
0 |
|
2 |
120 |
60 |
200 |
20 |
|
3 |
200 |
20 |
250 |
0 |
|
4 |
250 |
120 |
465 |
240 |
6.1.2 样品冷却后,在玛瑙研钵(4.7)中研磨,过45 μm标准筛(4.7),混匀,作为分析试样。研磨应采用边磨边过筛的方式,避免过度研磨造成石棉晶体形态或晶格受损,影响测试结果。
6.2 偏光显微镜检测
6.2.1 分析试样准备
取约20 mg分析试样(6.1.2)至15 mL离心管中,加入10 mL 20%甲酸溶液(4.4),加盖涡旋30 s,超声5 min,剧烈振摇后移取20 L至洁净的载玻片(4.6)上,于恒温烘箱中50 °C烘干后盖上盖玻片(4.6),平行制备3份。向3份样品的盖玻片边缘分别滴加折射率为1.550、1.620、1.690的浸油(4.2),使粉体颗粒充分浸润和分散,作为待测样。
6.2.2 观测和判定
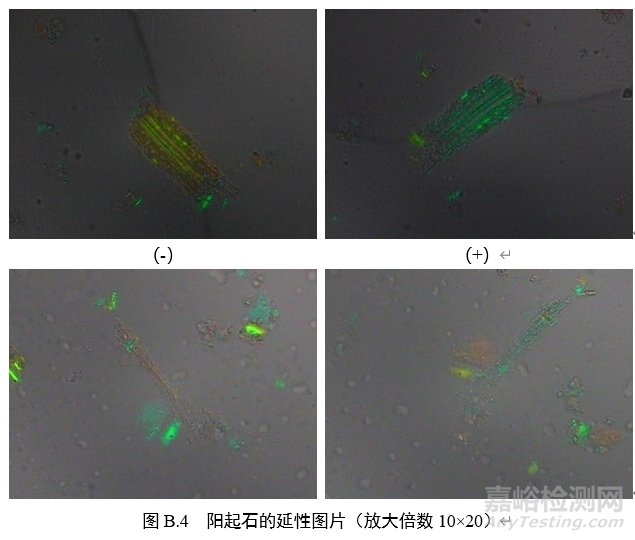
偏光显微镜使用10倍目镜,10~40倍物镜检测3份待测样,每份至少检测10个视野,不少于1000个颗粒。如3000个颗粒中累计发现4个或4个以上形态特征符合条件a或条件b的纤维颗粒,且存在光学特性符合附录表B.1的纤维,则转入扫描电子显微镜-能谱测定,否则判定为未检出石棉。
条件a:长径比大于20或长于5 µm的细针状纤维;
条件b:以下特征符合2项或以上:
1成束状的平行纤维;
2纤维束末端呈发散性;
3薄针状纤维;
4由单个纤维状物缠结而成的团块或弯曲状纤维。
6.3 扫描电子显微镜-能谱检测
6.3.1 仪器参数
扫描电镜工作电压:5 kV,工作电流:10 μA;
扫描电镜工作距离:8 mm;
能谱工作电压:15 kV;
能谱工作距离:15 mm。
6.3.2 分析试样准备
取6.2.1中剧烈振摇后的混悬液20 μL,滴于贴有导电胶(4.7)的扫描电镜观测台上,50 ℃烘干后作为待测样。平行制备3份。
6.3.3 检测和判定
在200~10000倍放大倍数下检测3份待测样,每份至少观测20个视野,不少于1000个颗粒。若3000个颗粒中累计观测到4个或4个以上形态特征符合6.2.2中条件a或条件b的纤维颗粒,则使用能谱分析纤维颗粒的元素组成。若能谱检出的元素种类与标准物质/标准样品一致(见附录表A.1、C.2),则判定检出石棉并报告种类。否则判定为未检出石棉。
7 分析结果的表述
7.1 检出浓度
以阴性分析试样(6.1.2)为基质进行加标实验,结果显示:
按6.2和6.3的方法检测,偏光显微镜和扫描电镜-能谱对于6种石棉的检出浓度均为0.1%。
实验室在检测时应随行使用阳性质控,确认能够满足检出浓度的要求。
7.2. 分析结果
对于偏光显微镜或/和扫描电镜-能谱未检出石棉的样品,结果表述为“未检出石棉”。
对于偏光显微镜和扫描电镜-能谱检出石棉的样品,结果表述为“检出…石棉”。
8 检测路径
本方法的检测路径如图1所示。
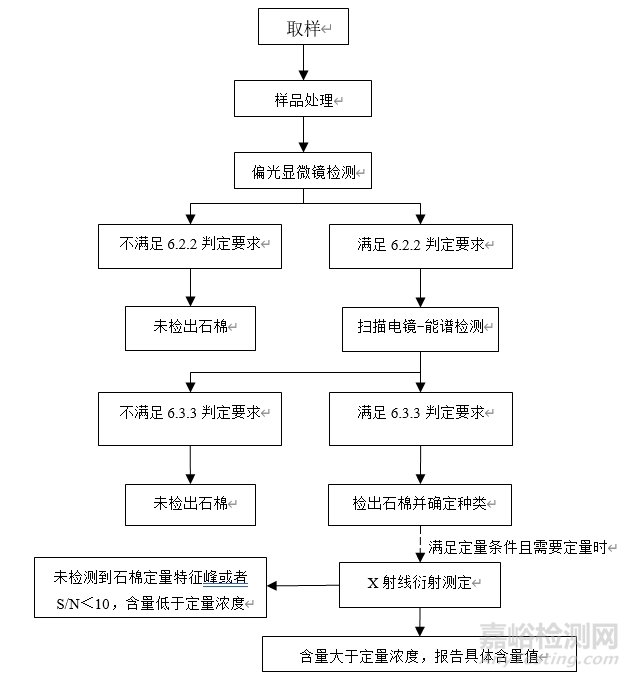
图1 化妆品中石棉检测的路径
附录A
(规范性附录)
滑石粉和各类型石棉的基本信息
表A.1 滑石粉和各类型石棉的基本信息
|
中文名称 |
英文名称 |
CAS号 |
化学式 |
|
滑石粉 |
Talc |
14807-96-6 |
Mg3(Si4O10)(OH)2 |
|
温石棉 |
Chrysotile |
12001-29-5 |
Mg3Si2O5(OH)4 |
|
青石棉 |
Crocidolite |
12001-28-4 |
Na2(Mg∙Fe)5Si8O22(OH)2 1 |
|
铁石棉 |
Amosite |
12172-73-5 |
(Fe∙Mg)7Si8O22(OH)2 2 |
|
直闪石 |
Anthophyllite |
77536-67-5 |
(Mg∙Fe)7Si8O22(OH)2 3 |
|
阳起石 |
Actinolite |
77536-68-6 |
Ca2(Mg∙Fe)5Si8O22(OH)2 4 |
|
透闪石 |
Tremolite |
77536-66-4 |
Ca2(Mg∙Fe)5Si8O22(OH)2 4 |
注:1. Fe/Mg≈4.5/0.5 (mol/mol)。
2. Fe/Mg≈4.5/2.5 (mol/mol)。
3. Fe/Mg<1/6 (mol/mol)。
4. 透闪石与阳起石的Mg/Fe浮动较大,且各类检测手段均不能很好区分二者。如有检出,种类可归为“透闪石/阳起石”。
附录B
(资料性附录)
偏光显微镜
B.1 偏光显微镜的技术要求
B.1.1 带有蓝色滤光片的光源,光源侧配备偏振镜或起偏器,偏振方向可调整至与检偏镜正交。
B.1.2 带有可360°旋转的载物台,旋转角度可以测量。
B.1.3 10~100倍物镜。
B.1.4 带有十字线的聚焦目镜,倍数为10~15倍。
B.1.5 配有照相装置,并可与电脑连接。
B.1.6 石膏试板。
B.1.7 λ补偿板或补偿器。
B.2 偏光显微镜的观测指标
表B.1 偏光显微镜下6种石棉纤维光学特性
|
石棉种类 |
单偏光下观测 |
正交偏光下观测 |
||
|
颜色1 |
多色性 |
消光2 |
延性符号3 |
|
|
温石棉 |
无色 |
无 |
平行 |
正延性+ |
|
铁石棉 |
无色至棕色 |
弱中 |
通常是平行 |
正延性+ |
|
青石棉 |
蓝色至棕灰色 |
强 |
通常是平行 |
负延性- |
|
直闪石 |
无色至亮棕色 |
无 |
平行 |
正延性+ |
|
透闪石、阳起石 |
无色至浅绿色、黄绿色 |
无或弱 |
近平行(约16°),混合纤维显示平行消光 |
正延性+ |
注:1.颜色和多色性:在单偏光模式下转动载物台,纤维颗粒从平行于偏光方向至垂直于偏光方向过程中呈现的颜色。
2.消光:插入检偏镜,在正交偏光模式下将载物台转动360°,纤维颗粒呈现四次明亮、四次黑暗特征。在完全黑暗时纤维颗粒的延长方向与目镜十字线的纵轴或者横轴之间的锐角夹角为纤维的消光角。
3.延性符号:在正交偏光镜模式下插入石膏试板,转动载物台至纤维颗粒的延长方向与目镜十字线呈45°(NE-SW),此时,纤维颗粒如呈现蓝绿色,则为正延性(+),如呈现橙色或者黄色,则为负延性(-)。
B.3 偏光品中石棉检测的路径
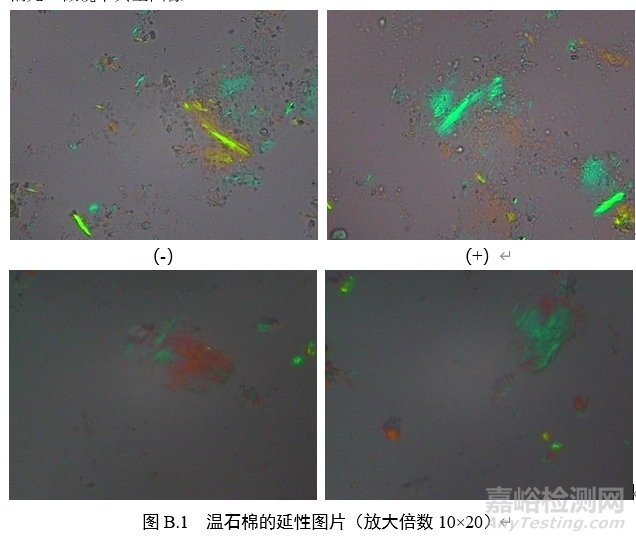
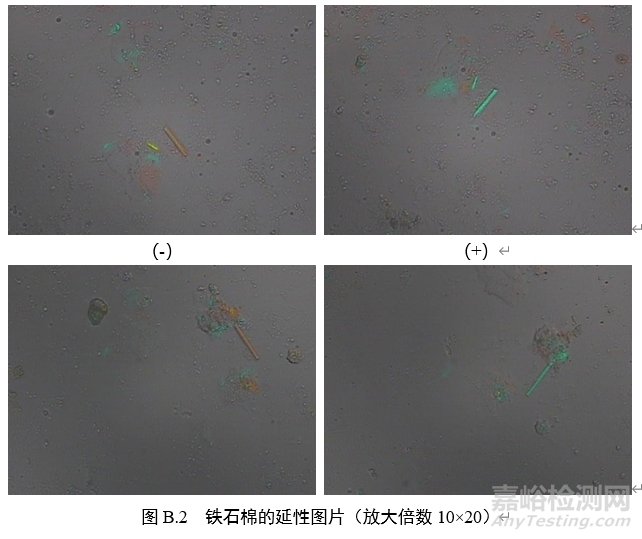



附录C
(资料性附录)
扫描电子显微镜
C.1 扫描电子显微镜的技术要求
C.1.1 加速电压:0.5~20 kV,放大倍数25~50000。
C.1.2 二次电子分辨率:1.0 nm(15 kV),2.0 nm(1 kV)。
C.1.3 能谱:能量分辨率<130 eV。
注:达到上述性能指标的电子探针显微分析仪也可代替扫描电子显微镜,用于本标准的分析。
C.2 能谱的观测指标
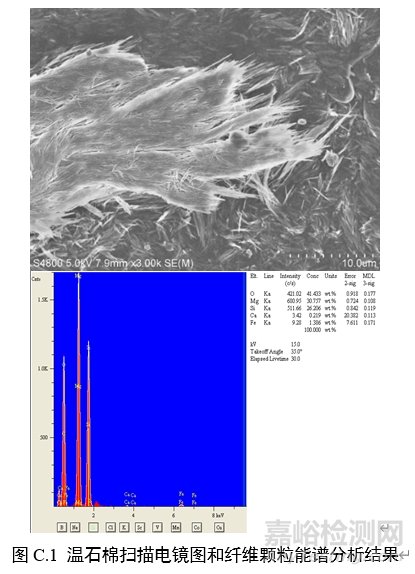
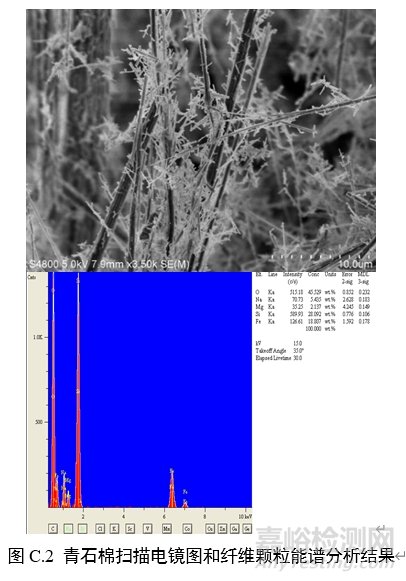
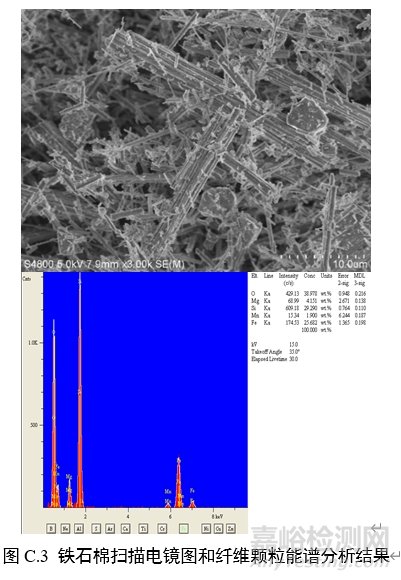
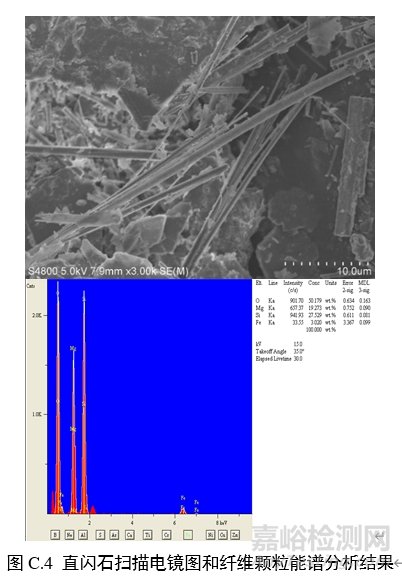
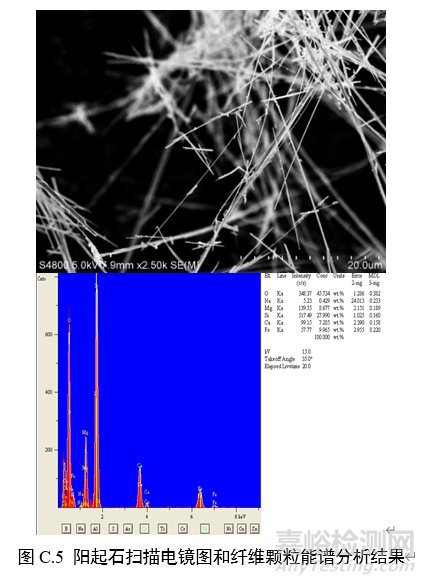
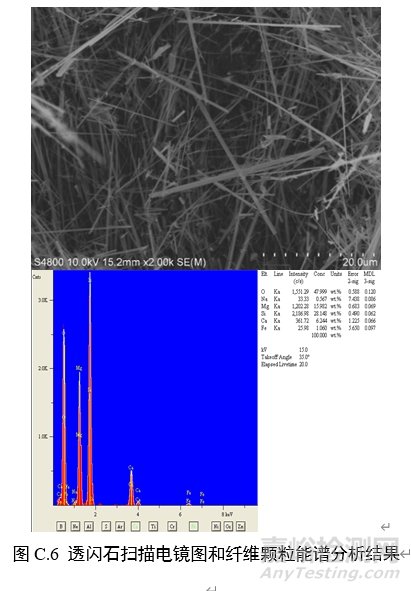
对纤维颗粒部位进行能谱分析,结果应含有石棉化学式中所有元素峰(不计氢),且无成峰型的铝、钾峰。扫描电子显微镜下各类石棉的典型图像和能谱分析结果见图C.1~图C.6。
附录D
(资料性附录)
石棉参考定量方法——X射线衍射仪测定法
5种角闪石石棉的主要特征衍射峰接近但吸收强度不同,当定性检出石棉且角闪石石棉种类不超过1种时,具备使用X射线衍射仪定量的条件。需要时,可进行定量测定。
D.1 X射线衍射仪的技术要求
D.1.1 多晶X射线衍射仪。
D.1.2 测角仪测角准确度优于0.02°(2θ)。
D.1.3 仪器分辨率优于60%。
D.1.4 综合稳定率优于±1%。
D.2 仪器参数
靶类型:Cu-Ka;
工作电压:40 kV;
工作电流:40 mA;
发散狭缝:1 mm;
散射狭缝:1 mm;
接受狭缝:0.3 mm;
扫描速度:1/16°(2θ)/min或20 s/步;
采样步宽:0.02°(2θ)/步;
测定扫描范围(2θ):10°~13°;34.5°~36°。
D.3 参比强度(K值)的测定
D.3.1 材料
6种石棉的K值各不相同,应根据定性检测中确定的石棉种类,选择相应的石棉标准物质/标准样品。选用的标准物质/标准样品应结晶良好,研磨至粒径小于0.040 mm。选用刚玉作为内标。石棉标准物质/标准样品和刚玉(4.5)置于105 ℃恒温烘箱内2 h,冷却至室温后使用。
D.3.2 制样
按1:1质量比,称取某种石棉标准物质/标准样品和刚玉(4.5)适量,放入玛瑙研钵(4.7)中研磨,形成均匀的混合试样。采用背压法制片,将样品框置于平板玻璃上,装入混合试样,用光滑的玻璃表面垂直压制成型,要求压力适中,表面平整,不存在波纹或凹痕。
D.3.3 衍射峰强度测量
将石棉标准物质/标准样品和刚玉混合试样压片置于X射线衍射峰上检测。每份混合试样重复制片测量5次。
D.3.4 计算K值
石棉的K值按式(1)计算:
Ki = Ii / Icor (1)
式中:
Ki ——i类石棉的参比强度;
Ii ——i类石棉的定量用衍射峰(见附录表D.1)的积分强度;
Icor——刚玉衍射峰的积分强度。
以5次测量的平均Ki值作为参比强度。
D.4 分析试样准备
称取约200 mg分析试样(6.1.2)至15 mL离心管中,加入10 mL 20%甲酸(4.4),加盖涡旋30 s,超声5 min,剧烈振摇后倾入洁净的蒸发皿(4.7)中,50 ℃烘干。
按1:1的质量比,精密称取上述分析试样和刚玉(4.5),在玛瑙研钵(4.7)中充分混合均匀,制作2份混合样品。采用背压法制片,将样品框置于平板玻璃上,装入混合样品,用光滑的玻璃表面垂直压制成型,要求压力适中,表面平整,不存在波纹或凹痕。
D.5 样品测定
于X射线衍射仪上检测压制的样品片,如采集的衍射图谱中含有石棉定量用特征峰(见表D.1),则按式(2)计算样品中对应石棉矿物的含量。
c=Ix*ms/(Is*mx*Ki)*100%/f (2)
式中:c——样品中石棉含量,%;
Ix——待测石棉衍射峰积分强度;
Is ——刚玉衍射峰积分强度,如存在本底衍射峰,应扣去本底值;
ms——刚玉的称样量,g;
mx——分析试样的称样量,g;
Ki——待测类型石棉的K值;
f ——样品灰化后的质量与灰化前质量的比值。
以2份平行样品结果的平均值作为最终测定结果。
表D.1 各类型石棉定量用衍射峰
|
石棉种类 |
2θ/(°) |
d/10-10m |
|
温石棉 |
12.01 |
7.36 |
|
铁石棉 |
10.60 |
8.34 |
|
青石棉 |
10.64 |
8.31 |
|
直闪石 |
10.61 |
8.33 |
|
阳起石 |
10.49 |
8.42 |
|
透闪石 |
10.55 |
8.38 |
注:阳起石和直闪石的最强衍射峰受滑石粉峰的干扰,因此定量用衍射峰均未选择其最强衍射峰。
D.6 定量分析结果的表述。
D.6.1 定量浓度
按D.2的仪器参数检测,X射线衍射法对于6种石棉的定量浓度均为1%。
D.6.2 分析结果
对于偏光显微镜、扫描电镜-能谱检出石棉,且含量达到X射线衍射定量浓度的样品,结果表述为“检出……石棉,含量为……”。
对于偏光显微镜和扫描电镜-能谱检出石棉,但X射线衍射未检测到石棉定量特征峰或者峰S/N<10的样品,结果表述为“检出……石棉,含量低于定量浓度”。
化妆品中葡糖醛酸等14种原料的检验方法
Determination of Glucuronic Acid andOther 13 Kinds ofComponents in Cosmetics
1 范围
本方法规定了高效液相色谱法测定化妆品中葡糖醛酸等14种原料的含量。
本方法适用于液体类、凝胶类、膏霜乳类化妆品中葡糖醛酸等14种原料含量的测定。
本方法所指的14种原料包括葡糖醛酸、酒石酸、羟基乙酸、苹果酸、乳酸、柠檬酸、2-羟基丁酸、乳酸甲酯、扁桃酸、乳酸乙酯、乳酸丁酯、二苯乙醇酸、柠檬酸三乙酯、羟基辛酸。
2 方法提要
以水提取化妆品中葡糖醛酸等14种原料,用高效液相色谱仪进行分离,二极管阵列检测器检测,以保留时间和紫外光谱图定性,峰面积定量,以标准曲线法计算含量。
本方法中14种原料的检出限、定量下限及取样量为1.0 g,定容体积为10 mL时检出浓度和最低定量浓度见表1。
表1 14种原料的检出限、定量下限和检出浓度、最低定量浓度
|
序号 |
原料名称 |
检出限 |
定量下限 |
检出浓度 |
最低定量浓度 |
|---|---|---|---|---|---|
|
(μg) |
(μg) |
(μg/g) |
(μg/g) |
||
|
1 |
葡糖醛酸 |
0.08 |
0.24 |
160 |
480 |
|
2 |
酒石酸 |
0.03 |
0.09 |
60 |
180 |
|
3 |
羟基乙酸 |
0.02 |
0.06 |
40 |
120 |
|
4 |
苹果酸 |
0.02 |
0.06 |
40 |
120 |
|
5 |
乳酸 |
0.05 |
0.15 |
100 |
300 |
|
6 |
柠檬酸 |
0.02 |
0.06 |
40 |
120 |
|
7 |
2-羟基丁酸 |
0.04 |
0.12 |
80 |
240 |
|
8 |
乳酸甲酯 |
0.04 |
0.12 |
80 |
240 |
|
9 |
扁桃酸 |
0.001 |
0.003 |
2 |
6 |
|
10 |
乳酸乙酯 |
0.04 |
0.12 |
80 |
240 |
|
11 |
乳酸丁酯 |
0.04 |
0.12 |
80 |
240 |
|
12 |
二苯乙醇酸 |
0.001 |
0.003 |
2 |
6 |
|
13 |
柠檬酸三乙酯 |
0.04 |
0.12 |
80 |
240 |
|
14 |
羟基辛酸 |
0.02 |
0.06 |
40 |
120 |
3 试剂和材料
除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T 6682规定的一级水。
3.1 磷酸氢二铵。
3.2 磷酸,优级纯。
3.3 甲醇,色谱纯。
3.4 乙腈,色谱纯。
3.5 甲酸,色谱纯。
3.6 0.05 mol/L的磷酸氢二铵溶液:称取6.60 g磷酸氢二铵,加水1000 mL溶解,用0.22 μm滤膜过滤。
3.7 标准品:14种原料标准品信息详见附录A。
3.8 葡糖醛酸等9种组分混合标准储备溶液
分别精密称取(精确到0.0001 g)葡糖醛酸、酒石酸、羟基乙酸、苹果酸、乳酸、柠檬酸、2-羟基丁酸、扁桃酸和二苯乙醇酸(3.7)适量于同一容量瓶中,加水适量,60 ℃超声15 min使溶解,用水定容,摇匀,配制成混合标准储备溶液备用,转入棕色储液瓶中,4 ℃避光保存,有效期1个月。标准储备溶液浓度见表2。
3.9 羟基辛酸单标标准储备溶液
精密称取(精确到0.0001 g)羟基辛酸(3.7)适量于容量瓶中,加水适量,60 ℃超声15 min使溶解,用水定容,配制成单标标准储备溶液备用,转入棕色储液瓶中,4 ℃避光保存,有效期1个月。标准储备溶液浓度见表2。
3.10 乳酸甲酯等3种组分混合标准储备溶液
分别精密称取(精确到0.0001 g)乳酸甲酯、乳酸乙酯、柠檬酸三乙酯(3.7)适量于同一容量瓶中,加水溶解并定容至刻度,摇匀,配制成混合标准储备溶液备用,转入棕色储液瓶中,4 ℃避光保存,有效期1个月。标准储备溶液浓度见表2。
3.11 乳酸丁酯单标标准储备溶液
精密称取(精确到0.0001 g)乳酸丁酯(3.7)适量于容量瓶中,加2 mL甲醇(3.3)溶解,用水定容,摇匀,配制成单标标准储备溶液备用,转入棕色储液瓶中,4 ℃避光保存,有效期1个月。标准储备溶液浓度见表2。
4 仪器和设备
4.1 高效液相色谱仪,二极管阵列检测器。
4.2 天平,感量分别为0.1 mg、0.01 mg。
4.3 超声波清洗器,功率100 W。
4.4 高速离心机,转速≥14000 r/min。
4.5 pH计,精度0.01。
4.6 涡旋振荡器。
5 分析步骤
5.1 混合标准系列溶液的制备
分别精密移取不同体积的葡糖醛酸等9种组分混合标准储备溶液(3.8)、羟基辛酸单标标准储备溶液(3.9)、乳酸甲酯等3种组分混合标准储备溶液(3.10)及乳酸丁酯单标标准储备溶液(3.11)于10 mL容量瓶中,用水定容。标准系列溶液现用现配。标准系列溶液浓度见表2。
表2 14种原料的标准储备溶液和标准系列浓度
|
原料名称 |
标准储备溶液浓度(mg/mL) |
混合标准系列溶液浓度(mg/mL) |
|||||
|---|---|---|---|---|---|---|---|
|
葡糖醛酸 |
8 |
0.1 |
0.2 |
0.4 |
0.8 |
2 |
4 |
|
酒石酸 |
4 |
0.05 |
0.1 |
0.2 |
0.4 |
1 |
2 |
|
羟基乙酸 |
8 |
0.1 |
0.2 |
0.4 |
0.8 |
2 |
4 |
|
苹果酸 |
8 |
0.1 |
0.2 |
0.4 |
0.8 |
2 |
4 |
|
乳酸 |
10 |
0.125 |
0.25 |
0.5 |
1 |
2.5 |
5 |
|
柠檬酸 |
10 |
0.125 |
0.25 |
0.5 |
1 |
2.5 |
5 |
|
2-羟基丁酸 |
10 |
0.125 |
0.25 |
0.5 |
1 |
2.5 |
5 |
|
乳酸甲酯 |
50 |
0.125 |
0.25 |
0.5 |
1 |
2.5 |
5 |
|
扁桃酸 |
0.2 |
0.0025 |
0.005 |
0.01 |
0.02 |
0.05 |
0.1 |
|
乳酸乙酯 |
40 |
0.1 |
0.2 |
0.4 |
0.8 |
2 |
4 |
|
乳酸丁酯 |
50 |
0.125 |
0.25 |
0.5 |
1 |
2.5 |
5 |
|
二苯乙醇酸 |
0.2 |
0.0025 |
0.005 |
0.01 |
0.02 |
0.05 |
0.1 |
|
柠檬酸三乙酯 |
50 |
0.125 |
0.25 |
0.5 |
1 |
2.5 |
5 |
|
羟基辛酸 |
50 |
0.125 |
0.25 |
0.5 |
1 |
2.5 |
5 |
5.2 样品处理
称取混合均匀的样品1.0 g(精确到0.001 g)于10 mL具塞比色管中,加水至10 mL刻度,充分涡旋混合30 s,60 ℃超声提取30 min,取适量样品在10000 r/min下高速离心15 min,取上清液过0.45 μm的滤膜,取续滤液作为待测溶液。如样品为油包水等难于在水中分散剂型,可先加入1 mL异丙醇涡旋混合30 s使分散均匀,再加水至10 mL刻度。
5.3 参考色谱条件
色谱柱:C18柱(250 mm×4.6 mm,5 μm),或等效色谱柱;
流动相:A:0.05 mol/L的磷酸氢二铵溶液(3.6),用磷酸(3.2)调pH值为3.0;
B:甲醇;
检测波长:214 nm;
柱温:室温;
进样量:5 μL。
表3 流动相梯度洗脱程序
|
时间/min |
流速/mL/min |
V(流动相A)/% |
V(流动相B)/% |
|
0 |
0.7 |
100 |
0 |
|
6.5 |
0.7 |
100 |
0 |
|
6.6 |
1.0 |
100 |
0 |
|
15 |
1.0 |
60 |
40 |
|
30 |
1.0 |
30 |
70 |
|
40 |
1.0 |
30 |
70 |
|
40.1 |
0.7 |
100 |
0 |
|
50 |
0.7 |
100 |
0 |
5.4 测定
在“5.3”色谱条件下,取混合标准系列溶液(5.1)分别进样,进行色谱分析,以标准系列溶液浓度为横坐标,峰面积为纵坐标,绘制标准曲线。
取“5.2”项下的待测溶液进样,根据保留时间和紫外光谱图定性,测得峰面积,根据标准曲线得到待测溶液中各原料的浓度。按“6”计算样品中各原料的含量。
6 分析结果的表述
6.1 计算
6.1.1 葡糖醛酸等10种α-羟基酸
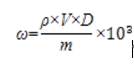
式中:ω——化妆品中葡糖醛酸等10种α-羟基酸原料的质量分数,μg/g;
ρ——从标准曲线得到待测原料的质量浓度,mg/mL;
V——样品定容体积,mL;
m——样品取样量,g;
D——稀释倍数(不稀释则取1)。
6.1.2 乳酸甲酯等3种乳酸酯类

式中:ω——化妆品中乳酸甲酯等3种乳酸酯类原料的质量分数(以乳酸计),μg/g;
m——样品取样量,g;
ρ——从标准曲线得到待测原料的质量浓度,mg/mL;
V——定容体积,mL;
D——稀释倍数(不稀释则取1);
90.08——乳酸的摩尔质量,g/mol。
6.1.3 柠檬酸三乙酯

式中:ω——化妆品中柠檬酸三乙酯的质量分数(以柠檬酸计),μg/g;
m——样品取样量,g;
ρ——从标准曲线得到待测原料的质量浓度,mg/mL;
V——定容体积,mL;
D——稀释倍数(不稀释则取1);
192.12——柠檬酸的摩尔质量,g/mol。
在重复性条件下获得的两次独立测试结果的绝对差值不得超过算术平均值的10%。
6.2 回收率和精密度
方法回收率为85%~115%,相对标准偏差小于10%。
7 图谱
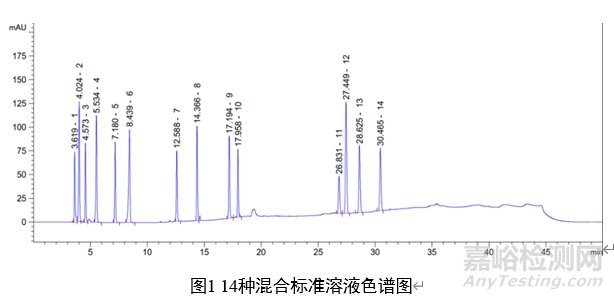
1:葡糖醛酸(3.619 min);2:酒石酸(4.024 min);3:羟基乙酸(4.573 min);4:苹果酸(5.534 min);5:乳酸(7.180 min);6:柠檬酸(8.439 min);7:2-羟基丁酸(12.588 min);8:乳酸甲酯(14.366 min);9:扁桃酸(17.194 min);10:乳酸乙酯(17.958 min);11:乳酸丁酯(26.831 min);12:二苯乙醇酸(27.449 min);13:柠檬酸三乙酯(28.625 min);14:羟基辛酸(30.465 min)
附录A
(资料性附录)
葡糖醛酸等14种原料标准品信息表
表A.1 葡糖醛酸等14种原料标准品信息表
|
序号 |
中文名称 |
英文名称 |
CAS登录号 |
分子式 |
相对分子质量 |
|
1 |
葡糖醛酸 |
Glucuronic acid |
6556-12-3 |
C6H10O7 |
194.14 |
|
2 |
酒石酸 |
Tartaric acid |
87-69-4 |
C4H6O6 |
150.09 |
|
3 |
羟基乙酸 |
Glycolic acid |
79-14-1 |
C2H4O3 |
76.05 |
|
4 |
苹果酸 |
Malic acid |
97-67-6 |
C4H6O5 |
134.09 |
|
5 |
乳酸 |
Lactic acid |
79-33-4 |
C3H6O3 |
90.08 |
|
6 |
柠檬酸 |
Citric acid |
77-92-9 |
C6H8O7 |
192.12 |
|
7 |
2-羟基丁酸 |
2-Hydroxybutyric Acid |
600-15-7 |
C4H8O3 |
104.10 |
|
8 |
乳酸甲酯 |
Methyl lactate |
547-64-8 |
C4H8O3 |
104.11 |
|
9 |
扁桃酸 |
Mandelic acid |
90-64-2 |
C8H8O3 |
152.15 |
|
10 |
乳酸乙酯 |
Ethyl lactate |
97-64-3 |
C5H10O3 |
118.13 |
|
11 |
乳酸丁酯 |
Butyl lactate |
138-22-7 |
C7H14O3 |
146.21 |
|
12 |
二苯乙醇酸 |
Benzilic acid |
76-93-7 |
C14H12O3 |
228.24 |
|
13 |
柠檬酸三乙酯 |
Triethyl citrate |
77-93-0 |
C12H20O7 |
276.28 |
|
14 |
羟基辛酸 |
Hydroxycaprylic acid |
617-73-2 |
C8H16O3 |
160.21 |
注:标准品可能存在不同形式,其CAS号也会不同,当与目标成分不同时需进行必要的折算。
附录B
(规范性附录)
葡糖醛酸等14种原料阳性结果的确证
因α-羟基酸极性大,紫外吸收接近截止波长,测定时易出现干扰,必要时可采用液相色谱-质谱法确证结果,以排除基质中其他组分的干扰。在相同的液相色谱-质谱实验条件下,如果样品中色谱峰的保留时间和紫外光谱图与标准溶液中对应成分一致(相对偏差在±2.5%之内),所选择的监测离子对的相对丰度比与相当浓度标准溶液的离子对的相对丰度比的偏差不超过表B.1规定范围,则可以判定样品中存在对应的测定成分。
表B.1 结果确证时相对离子丰度比的最大允许偏差
|
相对离子丰度(k) |
k>50% |
50%≥k>20% |
20%≥k>10% |
k≤10% |
|
允许的最大偏差 |
±20% |
±25% |
±30% |
±50% |
B.1 仪器参考条件
B.1.1 色谱条件
色谱柱:C18柱(100 mm×2.1 mm,1.8 μm),或等效色谱柱;
流动相:A:乙腈(含0.1%甲酸);B:0.1%甲酸水溶液;
表B.2 流动相梯度洗脱程序
|
时间/min |
V(流动相A)/% |
V(流动相B)/% |
|
0 |
0 |
100 |
|
4 |
0 |
100 |
|
15 |
95 |
5 |
|
17 |
95 |
5 |
|
17.1 |
0 |
100 |
|
22 |
0 |
100 |
流速:0.2 mL/min;
柱温:30 ℃;
进样量:5 μL。
B.1.2 质谱条件
离子源:电喷雾离子源(ESI源);
监测模式:正、负离子多反应监测模式;监测离子对及相关电压参数设定见表B.3;
鞘气流速:11 L/min;
干燥气流速:14 L/min;
鞘气温度:350℃;
干燥气温度:230℃;
碰撞气:Ar,20 psi;
离子源电压(IS)电压:正离子:4000 V;负离子:3000 V。
表B.314种原料的监测离子对及相关电压参数设定表
|
序号 |
组分名称 |
母离子(m/z) |
子离子(m/z) |
CE(V) |
|
1 |
葡糖醛酸 |
193.2 |
113.1* |
-14 |
|
131.1 |
-11 |
|||
|
2 |
酒石酸 |
149.1 |
73.0* |
-14 |
|
87.1 |
-18 |
|||
|
3 |
羟基乙酸 |
75.2 |
47.1* |
-16 |
|
45.1 |
-15 |
|||
|
4 |
苹果酸 |
133.2 |
115.0* |
-16 |
|
71.2 |
-17 |
|||
|
5 |
乳酸 |
89.2 |
43.1* |
-13 |
|
45.1 |
-15 |
|||
|
6 |
柠檬酸 |
191.1 |
111.1* |
-12 |
|
87.1 |
-11 |
|||
|
7 |
2-羟基丁酸 |
103.2 |
57.1* |
-13 |
|
45.1 |
-16 |
|||
|
8 |
乳酸甲酯 |
104.9 |
45.0* |
9 |
|
33.0 |
15 |
|||
|
9 |
扁桃酸 |
151.2 |
107.1* |
-15 |
|
76.9 |
-23 |
|||
|
10 |
乳酸乙酯 |
118.9 |
45.0* |
17 |
|
47.1 |
11 |
|||
|
11 |
乳酸丁酯 |
147.3 |
45.1* |
14 |
|
63.0 |
8 |
|||
|
12 |
二苯乙醇酸 |
227.1 |
183.2* |
-15 |
|
121.2 |
-9 |
|||
|
13 |
柠檬酸三乙酯 |
299.2 |
210.7* |
13 |
|
178.7 |
15 |
|||
|
14 |
羟基辛酸 |
159.2 |
113.2* |
-16 |
|
45.0 |
-17 |
*为推荐的定量离子。
注:柠檬酸三乙酯母离子为加钠离子。当柠檬酸三乙酯母离子为加氢离子时,参考母离子为277.1 m/z,子离子为157.0* m/z、203.1 m/z。CE为15 V、12 V。当采用不同质谱仪器时,仪器参数可能存在差异,测定前应将质谱参数优化。

来源:国家药监局


