您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-05 08:31
摘要
比较美国和欧盟的药品沟通交流制度和运行方式,为完善我国药品沟通交流制度提出建议。检索和梳理美国、欧盟的创新药、复杂仿制药、孤儿药、生物类似药等药品的沟通交流制度,分析我国药品沟通交流制度的实施现状并提出相应建议。 美国和欧盟根据药品研发生命周期和产品特性建立了针对不同类型药品的沟通交流会议,并积极与国际上其他监管机构和利益相关方共同开展沟通交流,助力新药研发。 相较而言,我国现有药品沟通交流会议类型对产品特性的针对性略显薄弱,沟通交流的答复仍有不足,还需积极寻求国际合作的机会。 我国沟通交流制度尚无法完全满足申请人的药品研发指导需求,借鉴国外沟通交流制度的经验和思路,我国应细化沟通交流会议的类型,尝试与国际其他监管机构达成审评合作,提高沟通交流的答复质量和透明度,提高药品的研发和审评效率。
【关键词】沟通交流制度;药品注册;科学建议;政策法规
药品沟通交流制度为制药企业与监管机构搭建了在新药研发和注册过程中消除信息不对等、达成共识的平台。在我国沟通交流指在药物研发与注册申请技术审评过程中,申请人与国家药品监督管理局药品审评中心(以下简称CDE)审评团队,就现行药物研发与技术指南不能涵盖的关键技术等问题所进行的沟通交流[1]。但在实践过程中,申请人存在有需求却不沟通、无效沟通或低效沟通、反复沟通或咨询、关键的研发决策问题相对较少等,与创新研发策略相适应的研发团队有待深入培养等问题[2-3],影响沟通交流以及药品研发的效率。
为降低和规避药物研发风险,切实提高药品研发注册上市的成功率,美国、欧盟等国家和地区均出台了沟通交流制度。我国2020年新修订的《药品注册管理办法》也将沟通交流制度纳入药品注册管理的基本制度,美国、欧盟的沟通交流机制可以为我国改进和完善相关制度提供宝贵经验。
1、美国药品沟通交流制度
美国FDA药品审评人员通过与申请人的沟通交流,对其提供药品的开发、审评以及上市许可申请相关的指导,形式包括面对面会议、电话会议和仅书面答复(written response only,WRO)。美国FDA针对创新药和生物制品、仿制药、生物类似药、非处方药分别制定了沟通交流的行业指南,规范沟通交流时间的合理安排,提高沟通交流效率,从而促进高效和有效的药物开发。下面针对各类会议的主要内容和沟通交流会议的流程分别进行概要阐述。
1.1 PDUFA会议
《处方药使用者付费法案》(Prescription Drug User Fee Act,PDUFA)建立了在创新药研究开发和审评过程中美国FDA与申请人沟通交流的正式会议制度,设置了A类会议、B类会议、B类[阶段结束(end of phase,EOP)]会议、C类会议、D类会议和初步监管建议(initial targeted engagement for regulatory advice on CBER/CDER producTs,INTERACT)会议。《FDA与PDUFA产品的申请人之间的正式会议》为美国FDA与创新药和生物制品的申请人之间的正式会议提供了行业指南规范[4]。以上6种会议的类型、时限和适用情形见表1。
▲表1-PUDUFA会议类型、时限和适用情形
1.2 GDUFA会议
《仿制药使用者付费法案》(Generic Drug User Fee Amendments,GDUFA)要求,美国FDA承诺为仿制药申请人在递交复杂仿制药申请前提供沟通交流等支持。《GDUFA下FDA与复杂制剂简略新药申请(abbreviated new drug application,ANDA)申请人之间的正式会议》[5]为美国FDA与复杂仿制药ANDA申请人之间的正式会议提供了途径。
GDUFA会议分为产品开发会议(product devel⁃opment meetings)、提交前会议(pre⁃submission meetings)、中期审评会议(mid⁃cycle review meetings,MCRM)、加强的中期审评会议(enhanced mid⁃cycle review meetings,EMCRM)以及完整回复函后的科学会议[post⁃complete response letter(CRL)scientif⁃ic meetings]5种。其中,对于MCRM和EMCRM而言,美国FDA在第1个评估周期中仅会批准其中1种会议申请,而不会同时批准这2种会议。GDUFA的会议类型、时限和适用情形见表2。
▲表2-GDUFA 会议类型、时限与适用情形
1.3 BsUFA会议
作为《生物类似药使用者付费法案》(Biosimilar User Fee Act,BsUFA)再授权的一部分,美国FDA承诺实现与申请者之间正式会议的管理目标。《FDA与BsUFA产品的申请人之间的正式会议》为美国FDA与生物类似药(或可互换生物制品)申请人之间的正式会议提供了行业指南规范[6]。
BsUFA会议分为生物类似药初步咨询(bio⁃similar initial advisory,BIA)会议、生物类似药开发(biosimilar biological product development,BPD)1~4类会议。BsUFA会议的类型、时限和适用情形见表3。
▲表3-BsUFA 会议类型、时限与适用情形
1.4 OMUFA会议
《非处方药专论使用者付费法案》(OTC Mono⁃graph Drug User Fee Program,OMUFA)承诺满足美国FDA和非处方(over⁃the⁃counter,OTC)药物申请人之间的会议申请,《FDA与非处方药专论产品的申请人之间的正式会议》为美国FDA与OTC专论产品的申请人之间的正式会议提供了行业指南规范[7]。
OMUFA会议分为X类会议、Y类会议和Z类会议。OMUFA会议的类型、时限和适用情形见表4。
▲表4-OMUFA会议类型、时限与适用情形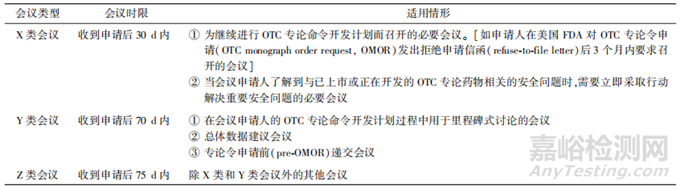
1.5 美国药品沟通交流制度的流程
PDUFA会议、GDUFA会议、BsUFA会议、OMU⁃FA会议的主要流程相同,见图1。不同会议的各阶段流程时限有所差异(见表1~表4),不在此进行展示。
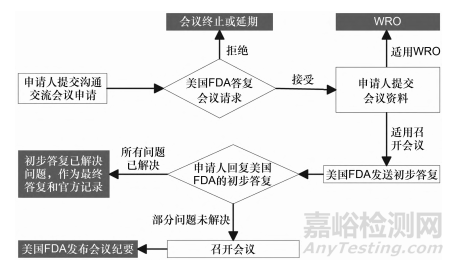
▲图1-美国沟通交流会议的流程
2、欧盟药品沟通交流制度
欧洲EMA的沟通交流制度主要包含科学建议和平行科学建议2种类型;还针对上市药物再利用项目、生物类似药、用于突发公共卫生事件的药物等分别设定其他科学建议情形。同时,将患者群体作为重要利益相关者纳入科学建议流程,可从监管者的视角为申请者在药物研发过程中遇到的问题提供建议。
2.1 科学建议
在药物的研发过程中,申请人可以要求欧洲EMA就最佳方法和研究设计提供指导,以形成关于药物安全性和有效性的可靠信息。作为药物监管生命周期中的重要工具,欧洲EMA为药物开发人员提供从制造过程质量管理到非临床研究和临床试验等(包括方法学问题)的开发计划的各个方面的科学建议。
欧洲EMA人用药品委员会(Committee for Medicinal Products for Human Use,CHMP)负责评估药品上市许可申请,它的作用之一是通过提供科学建议支持药物研究和开发。为实现该任务,CHMP成立了一个常设工作组———科学建议工作组(Scientific Advice Working Party,SAWP),为申请人提供科学建议(scientific advice,SA)和协议援助(protocol assistance,PA)。PA是获得孤儿药资格认定的申请人可以获得特殊形式的SA,其程序将主要遵循SA的程序,关注重点在于讨论临床数据是否可证实药品在上市许可阶段是效益显著的孤儿药状态。
2.1.1 科学建议的适用范围
欧洲EMA提供科学建议,以支持高质量、有效和安全药物的及时和合理的开发,并造福患者。科学建议的主要情形包括:①申请人正在开发一种创新药物,而欧盟指南、指导文件或药典中没有或缺少相关细节。②申请人正在开发针对病原体的新药或重新调整适应证的药物,这些病原体的医疗需求尚未得到满足,但缺乏或没有可用的指导原则。③申请人在其开发计划中选择偏离科学指南。④申请人对医药监管的了解有限,如一些学术团体或微型、小型和中型企业。
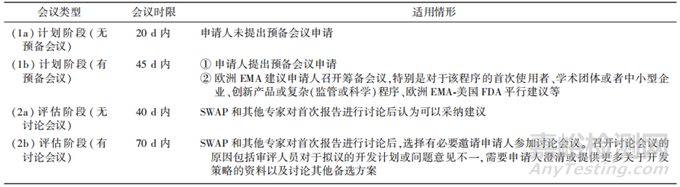
2.1.2 科学建议的程序和内容
科学建议的程序分为2个阶段:①有(45d)/无(20d)预备会议(preparatory meeting)的计划阶段。②有(70d)/无(40d)讨论会议的评估阶段,其类型、时限和适用情形见表5。
▲表5-科学建议的类型、时限和适用情形
2.1.2.1 计划阶段
申请人向欧洲EMA秘书处提交SA/PA申请并发送简报文件草案。自收到SA/PA申请起,欧洲EMA秘书处向SAWP提交申请,要求任命2名协调员。每个协调员组成一个评估小组,各自准备1份关于科学问题的报告和问题清单。欧洲EMA审评申请人提交的简报文件草案,并确认额外的专家/患者代表。申请人可以选择组织一次预备会议,若召开,则欧洲EMA将编写意见一览表(list of comments,LoC)并发送给申请人;若不召开,欧洲EMA可将意见以书面形式发送给申请人。最后,申请人根据相关意见修改并提交简报文件,欧洲EMA秘书处对最终简报文件进行验证。
2.1.2.2 评估阶段
协调员向欧洲EMA秘书处递交首次报告,SAWP同时咨询相关的欧洲EMA委员会、科学工作小组以及外部专家和患者,共同对首次报告的争议性问题进行讨论,并决定是否需要邀请申请人参加讨论会议。若召开,则SAWP向申请人反馈需要在讨论会议上解决的问题清单,申请人可在会议中提出问题清单范围外的讨论要点,协调员在会议结束时提出初步结论,并向SAWP反馈该会议的结果。最后,2名协调员完成联合报告,欧洲EMA秘书处起草最终建议函(final advice letter),SWAP采纳联合报告和建议函草案并由CHMP⁃SAWP/EMA对其进行同行评审,CHMP采纳最终建议函并反馈给申请人。相关流程见图2。
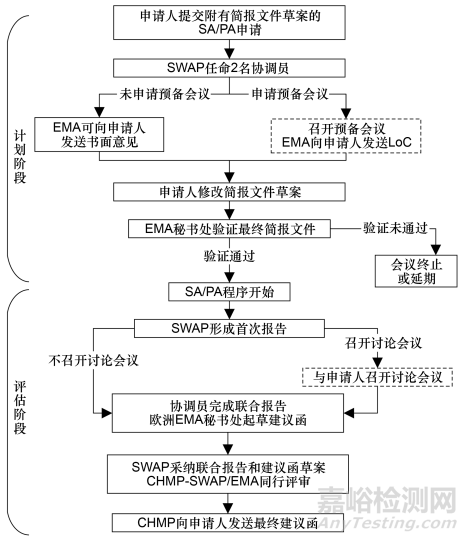
▲图2-欧洲 EMA 科学建议流程图
2.2 平行科学建议
欧洲EMA的平行科学建议包括EMA⁃FDA平行科学建议(EMA⁃FDA Parallel Scientific Advice,EMA⁃FDA PSA)和欧洲EMA与欧洲卫生技术评估网络21联盟平行科学咨询(Parallel EMA/EUnetHTA 21 Joint Scientific Consultation,EMA⁃EUnetHTA JSC),申请人可以同时获得多方的意见反馈。2种平行科学建议的时限和适用情形见表6。
▲表6-平行科学建议的类型、时限和适用情形
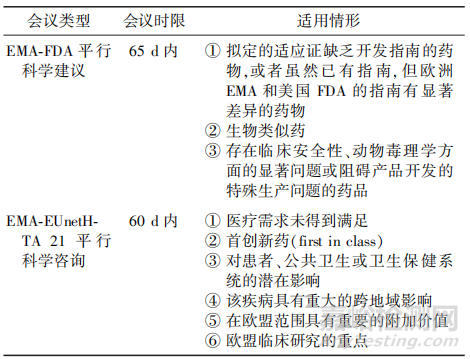
在EMA⁃FDA PSA计划中,欧洲EMA和美国FDA可共同在新药开发阶段与申请人就关键问题进行科学讨论和交换意见[9]。欧洲EMA和美国FDA关于药物研发问题的建议和上市许可的决定可能存在差异,将保留各自关于药物研发问题和上市许可的单独监管决策权。PSA作为同行讨论的一种方式,有利于药物开发方法与建议的趋同以及应对药物开发中的共同挑战,特别是在缺乏经验和棘手的科学问题领域。2021年,FDA⁃EMA复杂仿制药PSA试点启动[10],成为优化传统生物等效性方法的全球开发工具。PSA流程通常对应于欧洲EMA SAWP的70d科学咨询会议时间表和美国FDA的B类会议的时间表。首先,申请人向欧洲EMA和美国FDA提出PSA申请,经批准后,美国FDA和欧洲EMA分别进行内部讨论并交换初步意见,在程序开始后的第35d举行欧洲EMA/美国FDA双边会议,并在第65d举行申请人/欧洲EMA/美国FDA三边会议。
在EMA⁃EUnetHTA JSC中,欧洲EMA与EU⁃netHTA21提供平行科学咨询服务,其目的是使申请人可以获得监管机构和卫生技术评估(health technology assessment,HTA)机构关于证据产生计划的建议,以同时支持新药上市许可和报销的决策[11]。欧洲EMA和EUnetHTA21在程序开始前将举行行政电话会议,就会议主题进行初步交流;欧洲EMA、EUnetHTA21和申请人在程序d60共同参加F2F三边会议,重点关注监管机构和问题清单中提出的问题;在会议结束后,欧洲EMA和EUnetHTA21之间应举行一次闭门汇报,用于回顾、确定和讨论突出的分歧,再分别将最终建议给申请人[12]。
2.3 其他科学建议
欧洲EMA还针对上市药品再利用、生物类似药以及用于突发公共卫生事件相关疾病的药物制定了相关的科学建议指南,见表7。
▲表7-欧盟的其他科学建议的类型、时限、适用情形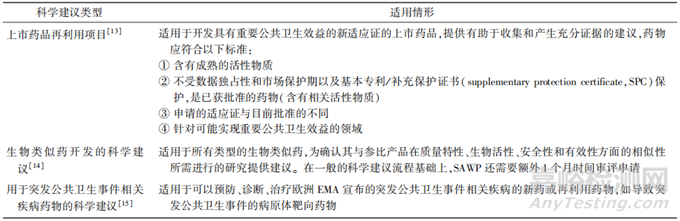
3、我国药品沟通交流制度
目前,CDE与申请人沟通交流和咨询的方式主要有召开沟通交流会议、一般性技术问题咨询、电话咨询、邮件咨询等。本文主要研讨沟通交流会议和一般性技术问题咨询。电话咨询和邮件咨询的作用较为简单,主要用于注册受理、审评业务的实时咨询以及与项目管理人沟通在审品种的日常管理,在此不再赘述。
3.1 沟通交流会议
2020年12月,国家药品监督管理局发布《药物研发与技术审评沟通交流管理办法》(2020年第48号通告),明晰了沟通交流的定义、类型、要求等事项,申请人可就现行药物研发与评价指南不能涵盖的关键性技术等问题与CDE开展面对面会议、视频会议、电话会议、书面回复等形式的沟通交流,充分阐述各自观点。我国沟通交流会议适用于中药、化学药和生物制品研发过程和注册申请技术审评中的沟通交流,分为Ⅰ类、Ⅱ类和Ⅲ类会议,仅对总体会议时限作出了规定,但未规定各阶段的时限。其会议类型、时限、适用情形见表8,基本流程见图3。
▲表8-我国沟通交流会议的类型、时限与适用情形

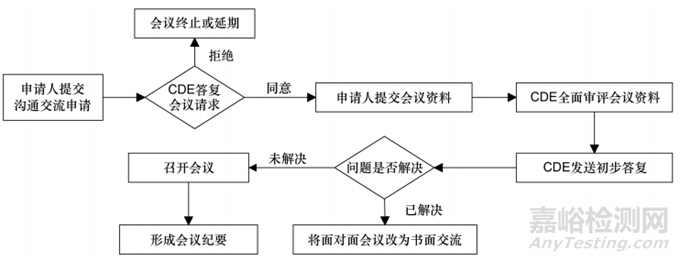
▲图3-我国沟通交流会议流程图
自2015年药品审评审批制度改革启动,我国已初步重建立起全新的、与国际接轨的审评审批制度和工作程序,药品沟通交流制度随之逐步完善。近年来,各类别的沟通交流申请数量一直逐年递增(见图4),反映出申请人在药品开发过程中对监管机构指导的迫切需求[22-23]。
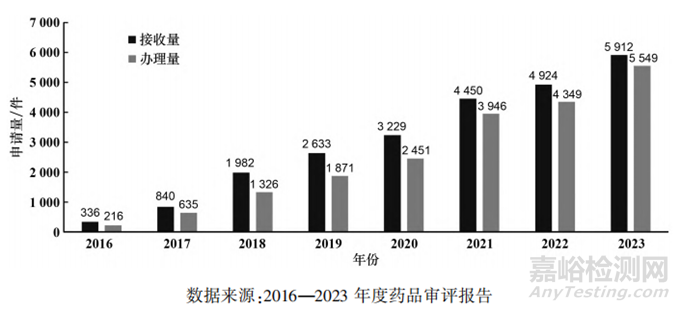
▲图4-2016-2023年CDE接收、办理及召开沟通交流会议申请量
3.2 一般性技术问题咨询
一般性技术问题包括与中心职能相关的技术审评问题及相关管理问题,不包括涉及保密的问题。CDE除了会答复申请人提出的一般性技术问题,也会公示受理共性问题和常见一般性技术问题。自2017年一般性技术咨询平台上线运行,其收到的一般性技术问题咨询数量总体呈现稳步增长,见图5。
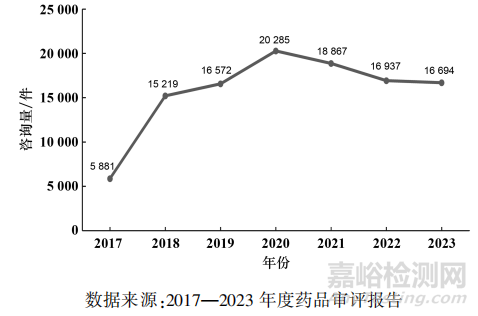
▲图5-2017-2023 年 CDE 接收一般性技术问题咨询量
4、完善我国药品沟通交流制度的建议
4.1 基于研发生命周期和产品特性细化沟通交流会议类型
目前我国的药品注册沟通交流会议类型与美国的创新药PDUFA会议类型基本一致。然而,美国FDA基于研发生命周期更加细分会议类型,体现了贯穿于新药研发生命周期各阶段的沟通需求。例如:美国FDA对B类会议进行了细化,将临床各期结束后会议作为一种特殊的B类会议,与其他B类会议的时限不同;并在新一轮付费法案下,引入了新的正式PDUFA会议类型,即D类会议和INTERACT会议,以提高沟通交流的效率和加强对药品早期开发阶段的支持。此外,美国FDA依据产品特性和不同付费相关法案,针对OTC、复杂仿制药、生物类似药等分别制定了对应的沟通交流会议指南;欧洲EMA则对孤儿药、上市药物再利用、生物类似药等制定相应的沟通交流程序。
相比之下,我国沟通交流会议虽然聚焦药物创新难点领域,就新型冠状病毒疫苗药物、罕见病用药、儿科用药等领域给予Ⅰ类会议或优先沟通交流的激励政策;但未真正基于各类药物研发特点及管理类别细化会议类型,也未明确对各类会议的具体要求。建议我国根据药品研发的生命周期和产品特性细分不同种类药品的会议类型,从而提高申请人和CDE之间沟通的效率和灵活性。例如:针对生物类似药、复杂仿制药、非处方药、罕见病用药建立与产品特性相适应的沟通交流制度,并针对新药设立适用于开发计划的早期沟通交流会议,赋能创新性产品的开发。
4.2 与多方监管机构建立合作
研究结果表明,FDA⁃EMA的平行科学建议有助于申请人优化药物开发并加深对监管决策的理解[16];EMA⁃EUnetHTA21的平行科学建议可以将监管和HTA观点整合到一个临床开发中,有利于研发者对药物收益⁃风险平衡以及附加价值的评估,加快落实药品在欧盟市场和国家市场的报销决策[17];欧洲EMA还邀请患者参与欧洲EMA科学咨询程序,患者通过提供对疾病及其治疗的真实体验来填补临床开发计划方面的信息空白[19]。随着中国加入ICH以及全球化趋势的影响,许多企业积极布局海外研发和注册申报。但目前我国的沟通交流机制中的主体仅涵盖CDE和申请人,申请人获取产品开发意见的渠道略为单一。建议我国参考欧美多方监管机构的互动形式,与其他国家药品监管机构加强合作,并根据不同国家或地区的要求,与其他国家的相关机构(如EUnetHTA21)达成审评合作,交换审评意见。此外,建议在沟通交流的过程中纳入患者代表、临床医生等利益相关方的意见,建设专家团队,以达到最大化灵活应用沟通交流制度的目的。
4.3 提高沟通交流的答复质量和透明度
欧洲EMA和美国FDA的药品沟通交流效率较高。欧洲EMA近3年均在规定时间范围内完成科学建议程序[20],2022年美国FDA对各类PDUFA会议在规定时间内响应的比例为89%~92%,在规定时间内开展的比例为66%~90%[21]。而我国存在沟通交流答复质量不够高、周期长等问题,由于CDE的审评资源和时间资源有限,难以达到申请人预期的沟通交流时间和答复质量[22]。建议加强审评人员的能力建设,更好地响应申请人的沟通交流需求;申请人应落实主体责任,主动学习相关法规和指导原则,提高沟通交流拟定问题和支持性材料的质量,从而实现高效交流。
此外,美国FDA和欧洲EMA均要求在药品上市审评报告中体现沟通交流的概况,而我国目前也在药品上市技术审评报告中公开了药品的沟通交流历史,但没有披露沟通交流的问题和结果。建议监管机构提高沟通交流环节的透明度,为同类型新药的研发和沟通交流提供参考,并帮助申请人更好地理解监管机构的立场和考虑因素。
5、结语
药品沟通交流制度为制药企业与监管机构搭建了在新药研发和注册过程中消除信息不对等、达成共识的平台,是我国深化药品审评审批制度改革中的重要实践。目前建立的双向多渠道、多层次沟通交流模式有助于降低和规避药物研发风险,提高药品审评审批质量和效率,在加速新药研发和上市中发挥积极作用。近年来,沟通交流机制随着相关法律法规和政策的出台而不断落实发展。在实践过程中,仍需结合我国国情并借鉴国外相关制度的经验完善这一制度,更好地指导申请人进行药物开发,顺应药物创新和研发全球化的趋势,满足公众的用药需求。
参考文献
[1] 国家药品监督管理局药品审评中心.国家药监局药审中心关于发布《药物研发与技术审评沟通交流管理办法》的通告(2020年第48号)[EB/0L].(2020-12-10)[2023-07-06.https ://www. cde. org.en/main/att/download/d3b07eeb5bc9908d0ac5493d31b56413.
[2] 杨建红,史继峰,温宝书,等.创新药研发与审评过程中的沟通交流[J].中国新药杂志,2010,19(19):1744-1746,1802.
[3] 韩玲,侯晨晨,中药新药研发与注册过程中的沟通交流[J]中国中药杂志,2021,46(3):730-735.
[4] FDA. Formal Meetings Between the FDA and Sponsors or Appli.cants of PDUFA Prducts EB/0L.(2023-09-22)[2023 -09-23 ]. https://www. fda. gov/media/172311/download.
[5] FDA. Formal Meetings Between FDA and ANDA Applicants ofComplex Products Under GDUFA Guidanee for Industry [ EB/0L].(2023-01-11)[2023-08-17]. https ://www. fda.gov/regulatory-information/search-fda-guidance-documents/for-mal-meetings-between-fda-and-anda-applicants-complex-produets-under-gdufa-guidance-industry.
[6] FDA. Formal Meetings Between the FDA and Sponsors or Appli-[3]ts of BsUFA Products Guidance for Industry[EB/0L].(2023-08 -09)[2023-08-07].https://www.fda. gov/regulatory-in-formation/seareh-fda-guidance-documents/formal-meetings-between-fda-and-sponsors-or-applicants-bsua-produets-guidance-industry.
[7]FDA. Formal Meelings Between the Food and Drug Administrationand Sponsors or Requestors of Over-the-Counter Monograph Drugs[EB/0L].(2022 -02-07)[2023-08 -17]. htps://www. fda.gov/regulatory-information/search-fda-guidance-documents/formal-meetings-between-food-and-drug-administration-and-sponsors-or-re-questors-over-counter.
[8]EMA,FDA. General Principles EMA-FDA Parallel SeientifieAdvice (Human Medicinal Prducts)[EB/0L].(2021 -08-18)[2023-06-11].https://www.ema. europa.eu/en/docu-ments/other/general-principles-european-medicines-ageney-food-drug-administration-parallel-scientific-advice_en. pdf.
[9]FDA, EMA. Pilot Prgram: EMA-FDA Parallel Scientifie AdviceFor Hybrid/Complex Generie Products-General Principles [ EB/0L].(2021-09-15)[2023-06-12].https://www. ema.europa.eu/en/documents/regulatory-procedural-guideline/pilot-programme-european-medicines-ageney-food-drug-administrationparallel-scientific-advice-hybrid/complex-generic-products-gener-al-principles_en. pdf.
[10] EMA. Parallel joint scientifie consultation with regulators andhealth technology assessment bodies[EB/0L].[2023 -06 -12 ].hitps ://www.ema.europa.euerhuman-regulatory/research-devel.opment/scientific-advice-pmtocol-assistance/parallel-joint-scientific-consultation-regulators-health-technology-assessment-bodies.
[11] EUnetHTA,EMA.Guidance on parallel EMA/EUnetHTA 21Joint Scientife Consultation[EB/0L].(2022-09-28)[2023-06-171.hitps://www.ema.europa.euen/documents/regulatory-procedural-guideline/guidance-parallel-ema/eunethta-21 -jointscientifie-consultation_en. pdf.
[12] EMA. Question and Answers on repurposing pilot pmjeet on proposal for framework to support not-for-prolit organisations and academiain repurposing authorised medicines[EB/0L].(2022-02-28)2023-06-18. https ://www.ema.europa.eu/en/documents/other/question-answers-repurposing-pilot-projeet-proposal-frame.work-support-not-profit-organisations_en. pdf.
[13] EMA. Tailored Scientifie Advice for biosimilar development: report on the experience from the pilot(2017-2020)[EB/0L].(2021-09-30)[2023-06-18].https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance#scientific-advice-on-biosimilars-section.
[14] EMA. Seientific advice and protocol assistance[EB/0L].[2023-06-18].https ://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance #f medicines.intended-for-a-disease-causing-public-health-emergency-section
[15 ]THOR S, VETTER T,MARCAL A, et al. EMA-FDA parallelscientifie advice: optimizing development of medicines in theglobal age[J]. Ther Innov Regul Sci,2023,57(4):656-661.
[16] TAFURIG,LUCAS I, ESTEVÃ0 S, et al. The impact of paral-lel regulatory-health technology assessment scientifie advice onclinical development. Assessing the uptake of regulatory andhealth technology assessment recommendations [J]. Br J ClinPharmacol.2018,84(5):1013-1019.
[17] EMA. Report of the pilot on parallel regulatory-health technologassessment scientific advice EB/0L].(2016-03-23)[2023-06-17.https://www.ema.europa.eu/en/documents/reportreport-pilot-parallel-regulatory-health-technology-assessment-sci-entifie-advice_en. pdf.
[18] MURPHY A,BERE N,VAMVAKAS S, et al. The added valueof patient engagement in early dialogue at EMA: seientifie adviceas a case study[」. Front Med,2021.8:811855.
[19] EMA. Annual activity report 2022[EB/0L].(2023-06 -07)[2023-12-071.https://www.ema.europa.ewen/documents/report/annual-activity-report-2022 en. pdf.
[20] FDA. FY 2022 PDUFA Performance Report[EB/OL].(2023 -04-07)[2023-12-07].https://www.fda.gov/media/166927/download.
[21]徐鹏遥,翟云,葛玉梅,等.以皮肤科药物为例谈我国药物研发沟通交流制度的改革与思考[J].中国新药杂志,2023,32(19):1941-1945.
[22]细胞和基因治疗产品临床相关沟通交流技术指导原则[J]中国医药导刊,2024.26(1):100-106.
[23]张思瑶,梁毅.欧盟和美国的平行科学建议制度对我国沟通交流制度的启示[J1.中国医药工业杂志,2023、54(8):1259 -1264

来源:中国新药杂志


