您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-07-14 21:25
近年来,以中国为代表的新兴医疗器械市场发展迅速,相应地审评对于医疗器械注册申报要求也越来越严格,其中原材料是医疗器械产品实现其预期功能的重要载体,也是器械安全性和有效性的重要保障。国家药监局审评中心希望注册申请人能够充分披露原材料信息,但是原材料生产商出于商业秘密的考量有保留地提供相关资料,基于此背景,主文档制度的出台是为了解决企业保护商业秘密的需求和审评机构要求对信息充分披露之间的矛盾。
2018年12月国家药监局为鼓励创新、优化注册申报流程,落实中共中央办公厅 国务院印发的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》精神,主文档中心项目组针对国内外主文档管理工作进行调研并结合我国实际情况发布了医疗器械主文档登记事项的4个相关文件,对外公开征求意见。征求意见稿是主文档相关文件对外征求意见稿一经发布即得到行业内的广泛关注。
2021年3月12日国家药监局正式发布《医疗器械主文档登记事项的公告》,与征求意见稿相比,调整了内容布局,逻辑条理更加清楚易懂;增加了《境内医疗器械主文档登记更新资料电子目录》;提示更新需提交全套主文档资料;取消纸质资料提交,明确申报形式与CA注册申报一致,已在系统中增加主文档端口。
主文档由其所有者自愿提交登记申请,监管机构对主文档进行登记。在登记时不经过实质审评,待关联医疗器械注册申请受理后一并审评。
主文档登记流程
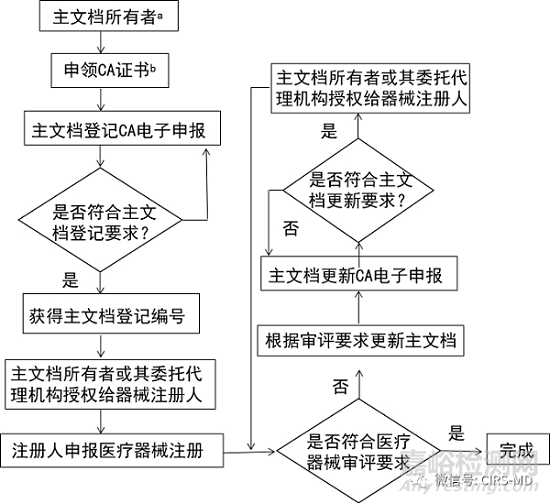

来源:CIRS医疗器械监管动态


