您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-12-23 20:14
无菌屏障系统是最终灭菌医疗器械安全性和有效的基本保证。本文介绍并比较了几种国内外常见的医疗器械包装材料微生物屏障性能的试验方法。并针对行业中讨论的材料微生物屏障试验的接受标准提出了意见和建议。
材料的微生物屏障特性评价方法
微生物屏障是在考虑了灭菌过程、搬运、运输、贮存等条件后所测试的无菌屏障系统防止微生物进入的性能。医疗器械常用无菌屏障系统包括不透气材料和透气材料。不透气材料比如塑料薄膜、塑料托盘、铝塑膜等,对于不透气材料,需要通过规范的试验方法证实材料是不透气,就意味着满足微生物屏障性能要求,标准ISO 11607-1附录 C(GB/T 19633.1 附录 A)给出了不透气材料阻气体通过的试验方法。对于透气材料(又称多孔性材料)的微生物屏障特性评价,目前国际上常见有三种测试方法:ASTM F1608(YY/T 0681.10)透气性包装材料微生物屏障分等试验,ASTM F2638(YY/T 0681.17)用气溶胶测试透气性包装材料微生物阻隔性能的标准测试方法,DIN 58953-6(YY/T 0681.14)灭菌医疗器械包装材料的微生物屏障试验。

我国医药行业标准YY/T 0681.10《无菌医疗包装试验方法第10部分:透气性包装材料微生物屏障分等试验》参考了ASTM F1608标准。该标准是在干态条件下使用枯草芽孢杆菌进行测试,用来测量透气无菌屏障材料阻止细菌孢子穿透的能力。
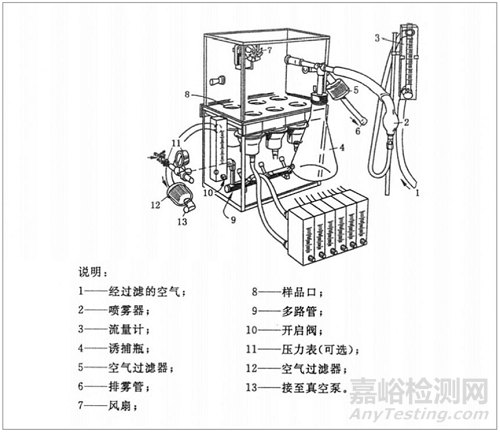
图1:试验箱
完全不穿透的对照样品(微生物穿透率为零)受到100万或10^6个菌落形成单位(cfu)的挑战,cfu 10^6的数量的log10值为6。如果以与对照相同的方式挑战的样品允许10cfu(log10=1)穿透,则其对数减少值(LRV)为5。因此,LRV越高,透气包装材料对微生物穿透的抵抗力就越强。表1显示LRV与相对应的芽孢截留率的关系。无菌屏障系统应尽可能选择较高 LRV 值的透气材料,以保证器械的无菌保证水平并减少无菌失效的风险。目前,该方法能够被行业内广泛接受,国内外开展该方法的实验室相对也较多。
该方法给出了精密度和偏倚的说明。该信息基于1993年ASTM进行的实验室协同研究。十一个实验室各由一个操作人员对从六种材料上分别随机裁取的两个样品和一个阳性对照进行试验。使用了汇总的所有材料的实验室内和实验室间的变异估计值来得到具有足够自由度的一个操作者和多实验室的精密度估计值。另外,由于 LRV 的真实值是不可知的,不能验证该方法的偏倚,所以该方法的偏倚不可知。
我国医药行业标准YY/T 0681.17《无菌医疗包装试验方法第17部分:透气包装材料气溶胶过滤法微生物屏障试验》参考了ASTM F2638标准。该标准使用1.0μm粒子的气溶胶作为替代微生物来测定透气包装材料屏障性能的标准测试方法,测量透气包装材料阻止颗粒穿透的能力。所有材料都具有出现最大穿透百分比(%PMax)的面速度。穿透百分比越低,性能越好。无菌屏障系统应尽可能选择较低%PMax值的透气材料,以保证器械的无菌保证水平并减少无菌失效的风险。目前,该方法能够被行业内接受,国内外开展该方法的实验室在逐步增加。
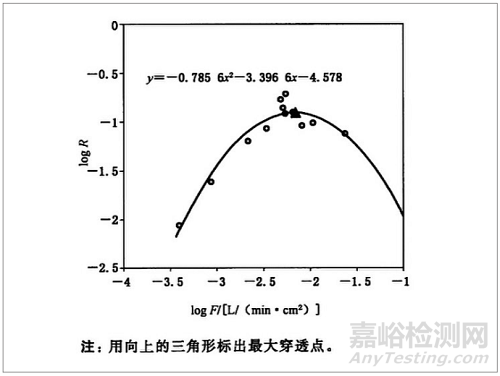
图 2:典型穿透 - 流量曲线
使用微生物测试透气包装材料的屏障性能与测量材料的过滤效率具有相关性。该试验方法不需要采用微生物学的方法,而采用材料过滤微粒的能力来评价材料的屏障性能。当测定透气包装材料的过滤效率是,可得到一条典型的过滤效率曲线(见图 2),特定材料的曲线的弧度取决于材料本身。最大穿透率可以作为评判材料屏障性能优劣的重要依据。

图 3:ASTM F2638 测试设备
该方法的精密度是基于美国材料试验协会2004年进行的单计数器法的实验室内研究和2006年进行的双计数器法的补充研究。试验测试设备见图3。在2012年的研究中,2个实验室在总共6个试验单元中分析了4种不同的透气包装材料。两个独立的实验室内试验结果证实,无论是单计数器还是双计数器配置,方法均具有重复性。在研究当时,没有用于确定本试验方法偏倚的适宜的可接受的标准物质,因此不给出偏倚的描述。
我国医药行业标准YY/T 0681.14《无菌医疗器械包装试验方法第14 部分:透气包装材料湿性和干性微生物屏障试验》参考了DIN 58953-6标准。该标准规定了湿性条件和干性条件微生物屏障实验的试验方法。湿性条件微生物屏障试验是将微生物液滴滴加到试验样品上。液滴干燥后,进行试验以测试是否有微生物穿透到试样样品的另一面。干性条件微生物屏障试验是通过对用待测密封的微生物屏障装置中的空气进行冷却,空气流将进入试验瓶中。如果冷却前包装材料上有微生物培养物覆盖,空气流可能会使携带微生物的颗粒穿过包装材料。用微生物学技术记录通过包装材料的任何微生物并进行评价。该试验方法被设计为“合格、不合格”型试验。该方法预先设定了一个接收准则,“如果全部10个试验样品生长的菌落数不超过15个,且任何1个试验样品生长的菌落数不超过5个,则认为包装材料足以作为无菌屏障”。目前没能找到DIN 58953-6标准设定包装材料屏障性能的接受准则为“15个和5个”菌落数理由和逻辑关系。
我国卫生部门发布的《消毒产品卫生安全评价规定》中规定,进入医疗机构的带有灭菌标识的灭菌物品包装物需进行微生物屏障试验,《消毒技术规范》给出了相应的试验方法和判定准则。该方法与《消毒技术规范》给出的试验方法和判定准则是一致的,可视为供给医疗机构灭菌的透气材料的基本要求之一。
针对以上三种常见的试验方法,将其进行了比较来说明每种试验方法存在的优劣。见以下表 2,可点击放大查看。

材料微生物屏障试验方法的讨论与建议
用于医疗器械灭菌的透气包装材料常提供给医疗机构和医疗器械制造商。由于两种情况下诸如器械的特点、灭菌方式、运输条件、贮存条件和贮存期限等的差异,按风险管理要求,对材料的微生物屏障性能提出不同的要求。
DIN 58953-6(YY/T 0681.14)给出的试验方法与我国消毒技术规范中的按照消毒产品进行管理的灭菌物品包装物的测试方法和接收准则是一致的。该方法需要微生物实验室,预先设定了接收准则,适合于医疗机构和使用者对灭菌透气包装材料的微生物屏障特性进行初步检查控制。该方法不能给出定量的数据,不能为选择透气包装材料提供定量的数据,无法对透气包装材料的一致性进行评价。
而对于医疗器械制造商而言,需要对透气包装材料的微生物屏障性能进行定量的评价,以确定包装材料是否足以提供屏障性能以应对各种挑战,如灭菌过程,运输、搬运和贮存过程的挑战。ASTM F1608(YY/T 0681.10)和ASTM F2638(YY/T 0681.17)给出的方法和结果,为使用者在无菌医疗器械的包装设计,包装验证等环节提供了帮助。ASTM F2638(YY/T 0681.17)方法无需微生物培养,模拟实际使用环境的挑战条件,能够较快速地检测透气材料的微生物屏障特性,适于医疗包装的生产商和包装使用厂商对包装材料的常规控制和选择比较。ASTM F1608(YY/T 0681.10)采用了 10^6 CFU / 样品微生物挑战量和较为极端的挑战风速(2.8L/min),评价包装材料的微生物屏障性能。这二种定量的方法,能按透气材料的屏障性能高低给出分等排序。这二种方法没有给接收准则。对于医疗器械制造商而言,接收准则需要根据医疗器械性能,临床应用,所经受不同灭菌过程、运输条件以及贮存条件来确定。
小结
GB/T 19633.1 中 5.2.3 规定“透气性材料应能提供适宜的微生物屏障,以提供无菌屏障系统的完整性和产品的安全性”。YY/T 0681.10和YY/T 0681.17提供了定量的微生物屏障试验方法,用于确定透气包装材料的微生物屏障特性。YY/T 0681.14是在假定接受准则的条件下,定性地评价透气材料的微生物阻隔性能。选择医疗器械无菌屏障系统时,应考虑产品在使用前无菌屏障系统维持无菌的能力。国内外法规越来越关注医疗器械的风险管理,无菌屏障系统在设计和开发阶段,需要尽可能降低风险,按 ISO 11607(GB/T 19633)要求,通过包装验证来选择具有足够微生物屏障性能的包装材料。
医疗器械包装微生物屏障性能测试方法探讨(摘选)
作者:朱雪燕、钱军
杜邦(中国)研发管理有限公司

来源:Internet


