您当前的位置:检测资讯 > 监管召回
嘉峪检测网 2022-01-28 21:58
如果说每一种事物都有它的培土播种、生根发芽、成长成熟、暮老病亡,盖棺定论等等阶段,那么一款医疗器械的整个生命周期中,也会它的生老病死。
为了更好地服务医疗器械的病老,政府也是花了很大力气的,据不完全统计,为服务好“病老”的医疗器械,政府近五年公布了至少12条相关的“治疗方案”,分列如下:
1. 医疗器械召回管理办法2017.05.01
2. 医疗器械不良事件监测和再评价管理办法2019.01.01
3. 国家药监局综合司、国家卫生健康委办公厅关于印发《医疗器械唯一标识系统试点工作方案》的通知2019.07.01
4. 医疗器械唯一标识系统规则2019.10.01
5. 国家药监局《关于做好第一批实施医疗器械唯一标识工作有关事项的通告》2019.10.12
6. 医疗器械注册人开展不良事件监测工作指南2020.04.03
7. 医疗器械定期风险评价报告撰写规范2020.06.30
8. 国家药监局、国家卫健委、国家医保局《关于深入推进试点做好第一批实施医疗器械唯一标识工作的公告》2020.09.29
9. 医疗器械注册人开展产品不良事件风险评价指导原则2020.11.25
10. 医疗器械注册人备案人开展不良事件监测工作检查要点2021.04.09
11. 医疗器械监督管理条例(2021) 2021.06.01
12. 国家药监局、国家卫健委、国家医保局《关于做好第二批实施医疗器械唯一标识工作的公告》2021.09.13
总体上来说,在这一部分,主要有三方面的控制管理措施,包括不良事件监测、产品召回、以及信息追溯。
一、不良事件监测:
在2019年元旦生效的《医疗器械不良事件监测和再评价管理办法》中,对于不良事件检测,给出了具体行之有效的要求及措施,具体包括四方面:
1.1:原则:秉持可疑即可报原则
1.2:义务:其使用单位、持有人、经营企业均对监测有主体义务。
1.3:归类:分为个例不良事件、群体不良事件。
1.4:流程:分为信息收集、报告、调查、分析、评价、控制等六大进程。
在收集阶段,其信息可以从多方面收集,如医疗器械经营企业、使用单位、使用者、或者是国家医疗器械不良事件监测信息系统等等。在报告阶段,经营企业以及使用单位可以向持有人报告,监测的义务主体可以向监管部门报告。在调查阶段,包括产品质量状况、伤害与产品的关联性、使用环节的操作过程、流通过程的合规性等等均在被调查之列。完成基础数据的调查后,分析事件发生的原因,然后评估产品的风险。最终,会有如下十种可能的控制措施:
1)停止生产、销售相关的产品
2)通知医疗器械经营企业、使用单位暂停销售和使用。
3)实施产品的召回。
4)发布风险信息
5)对生产质量管理体系进行自查、并对相关问题进行整改
6)修改说明书、标签、操作手册等
7)改进生产工艺、设计、产品技术要求等。
8)开展医疗器械再评价
9)投规定进行谈变更注册或备案
10)其它需要采取的风险控制措施
在2020年4月3日生效的《《医疗器械注册人开展不良事件监测工作指南》中,对医疗器械不良事件报告进行了详细的流程指引:
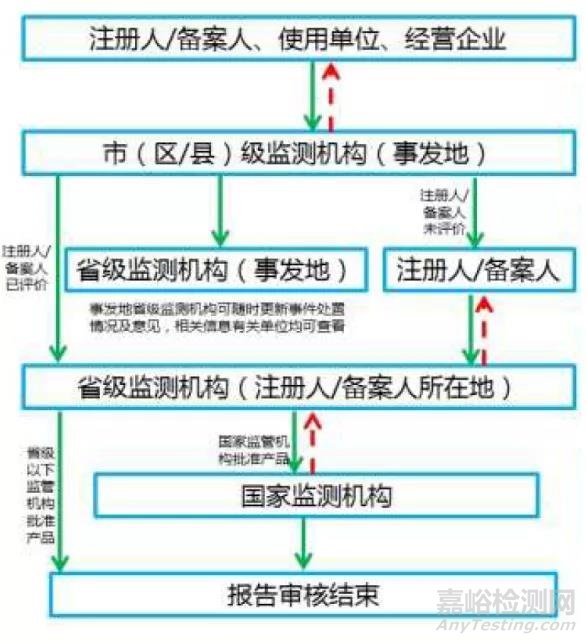
分五大步骤:
(一) 上报:死亡事件7日内报告,严重/可能导致严重伤害或死亡事件的20日内报告。
(二) 市级审核:10日内审核报告真实性、准确性、完整性,对审核不通过的,退回报告单位处理。
(三) 报告评价:注册人/备案人30日内完成死亡报告评价,45日内完成严重/可能导致严重伤害或死亡事件的报告评价,并且补充内容需随时提交。
(四) 省级审核:10日内审核注册人/备案人的评价结果,对审核不通过的,退回注册人/备案人。
(五) 复核:复核不通过的退回省级监测机构。
二、产品的召回
在这一部分,还是遵循2017年05月01日生效的《医疗器械召回管理办法》,分为十大部分:
2.1:召回的责任主体:境内医疗器械产品注册人或者备案人,进口医疗器械的境外制造厂商在中国境内指定的代理人。
2.2:召回的范围:存在缺陷的医疗器械产品。
2.3:缺陷范围包括三大部分:一是正常使用下有不合理安全风险。二是不符合强制性标准、经注册或者备案的产品技术要求。三是生产经营不符合医疗器械GMP,GSP导致可能存在不合理的风险。
2.4:召回可分为主动召回以及责令召回。
2.5:召回等级分为:一级召回、二级召回、三级召回。
2.6:召回责任主体必需履行的义务包括:建立健全召回管理制度,收集医疗器械安全相关信息,对可能的缺陷产品进行调查、评估、及时召回缺陷产品。
2.7: 对于医疗器械经营企业、使用单位必需履行的义务定义:积极协助召回责任主体对缺陷产品进行调查,评估、主动配合履行召回义务,并按照召回计划及时传送、反馈医疗器械召回信息、控制以及收回缺陷产品
2.8:产品召回信息发布媒介:一级召回由国家药监局网站和中央主要媒体发布,二级以及三级召回由省级药监局网站公布。
2.9:产品召回通知的对象为有关经营企业、使用单位或使用者。
2.10:召回主体提交监管部门文件内容包括:医疗器械召回事件报告表、调查评估报告、召回计划、召回记录、医疗器械召回总结评估报告等。
三、信息追溯:医疗器械唯一标识
这是一个比较新的内容,在新《条例》第三十八条中,要求:国家根据医疗器械产品类别,分步实施医疗器械唯一标识制度,实现医疗器械可追溯。这就涉及到一个系统,即医疗器械唯一标识系统,该系统的发展历程如下:
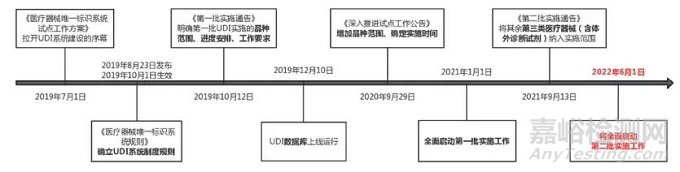
它是由医疗器械唯一标识(unique device identifier, UDI)、UDI数据载体和UDI数据库组成。其中UDI是指附载在医疗器械产品或者包装,由数字、字母或者符号组成的代码,用于对医疗器械进行唯一性识别。UDI应符合唯一性、稳定性和可扩展性的要求。UDI规则的实施步骤可分为“产品赋码—标识申报—数据共享—数据应用”四个环节。
根据上述发展历程,我们也隐约可以看出如下三信关键信息:
第一:在《第一批实施公告》《深入推进试点工作公告》规定的9大类69个品种的基础上,将其余第三类医疗器械(含体外诊断试剂)纳入第二批实施唯一标识范围,支持和鼓励其他医疗器械品种实施唯一标识;
第二:2022年6月1日起,纳入第二批实施范围的医疗器械应当具有医疗器械唯一标识,此前已生产可不具有唯一标识;
第三:医疗器械注册人负责开展唯一标识赋码、注册系统提交、数据库提交等相关工作。
回过头来,我们再看看这个医疗器械的生老病死,不难发现,社会在发展,在进步,在日新月异,制度也在发展,也在进步,在越来越完善。我们有理由相信,在这良好的设计、生产、经营、提升、更新淘汰的生长环境中,医疗器械的发展当会茁壮成长,为拯救及延续生命,改善病人生活、体验做出更多贡献。

来源:德大器械产业管家


