您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-03-31 22:35
摘要:2021年, 尽管新冠疫情仍然困扰全球, 但新药创制的脚步却未因此减缓。美国 FDA 药物评价和研究中心(CDER)在过去一年里共计批准了50款新药, 其中有 27款为首创性(first-in-class)药物, 为过去十年的峰值。小分子的首创性药物依旧占据主导地位, 获批15款。其中包括多个具有里程碑式重要意义的首创性小分子药物, 例如首个靶向“不可成药”靶标KRAS G12C 突变蛋白的小分子共价抑制剂索托雷塞(sotorasib), 首个靶向 BCRABL1 蛋白肉豆蔻酰口袋的小分子变构抑制剂阿思尼布(asciminib), 首个抑制缺氧诱导因子HIF-2α 的小分子抑制剂贝组替凡(belzutifan), 首个治疗慢性心力衰竭恶化的sGC小分子激动剂维立西呱(vericiguat)等。首创性药物依赖于发现全新的作用靶标和生物机制, 分子设计思路各不相同, 具有重要的学习和借鉴意义。本文通过浅析其中 3 例首创性小分子药物的研发背景、研发过程和治疗应用, 以期为更多的首创性药物提供研究思路与方法。
在过去的一年里, 虽然COVID-19 新冠疫情持续影响着人们的生活和工作, 造成了难以估量的损失, 但疫苗的接种、中和抗体和口服小分子抗病毒药物的研究也正在为我们的安全保驾护航。今年, 病毒的持续突变成为了新的难题, 也给药物的研究不断带来新的挑战。从人类对抗各类疾病的历史来看, 我们对生命体的深入理解还需要漫长的时间与更多的基础研究工作, 创新药物的研究也依赖于对生命体复杂机制的探索与剖析。药物研发的历史经历了如下几个关键阶段:①1899年至1970年, 这一阶段的药物研究只能发现药效物质, 明确药物的基本结构, 但作用机制未知(defined molecules, undefined molecular targets)。例如1899年前后明确了阿司匹林的解热镇痛作用和分子结构, 虽然不清楚其具体的作用机制, 但这也并不妨碍阿司匹林的广泛使用。直到1970年前后才逐步明晰了环氧合酶的作用机制, 阐明了阿司匹林的作用机制;②1970年至今, 这一阶段的里程碑事件是抗溃疡药物西咪替丁的成功研制。通过以内源性物质组胺为结构模板, 有目的地设计H2 受体拮抗剂, 开启了合理药物设计的先河。合理药物设计旨在明确分子与靶标的对应关系(defined molecules and defined molecular targets)并基于蛋白的空间结构使得药物设计的目标更加明确。时至今日, 绝大部分的药物研发案例均是基于药物靶标的结构而开展的。③2010年至今, 这一阶段的药物研究开始不局限于单一的药物靶标, 而是充分利用内源性的蛋白功能进行更加复杂的药物设计。例如当下研究火热的靶向蛋白酶体的分子嵌合体(Proteolysis Targeting Chimeras, PROTACs), 即是通过双功能分子特异性招募E3泛素连接酶并实现目标蛋白的选择性降解[1]。因此, 回顾以上不同的药物发现阶段, 研究方法虽在不断更新迭代, 但任何时期都需要具有全新作用机制的创新药物。
目前, 通常将新药创制的过程分类为首创性药物(first-in-class)和跟随性药物(me too, me better), 二者都对人类健康具有重要意义[2]。为了避免在热门靶标上的恶性竞争与资源浪费, 许多国家陆续更新了新药评审政策并要求跟随性药物需要与首创性药物进行“头对头”的对照实验, 这对跟随性药物的研发提出了更高的要求与挑战。在国内外新药创制大环境的驱动下, 首创性药物的研究正逐渐成为制药企业与科研院所追求的目标。首创性药物的研究是生物学驱动的, 需要大量的基础研究作为铺垫。其研究过程首先需要发现全新的功能蛋白及其致病机制, 随后通过化学小分子或生物大分子进行靶向干预, 不断深化靶标再验证的过程。在明确新靶标的安全性、有效性及其与疾病的因果性关系后, 再优化其活性、理化性质和成药性等, 最终才有望得到临床前的候选化合物。首创性药物的高风险、长周期、大投入让不少研究人员望而却步, 但成功后的高回报以及对人类健康的巨大推进作用, 都值得不断追求和探索[3-5]。
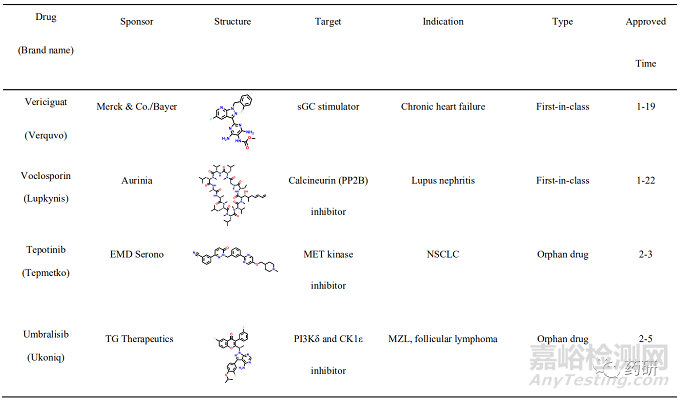
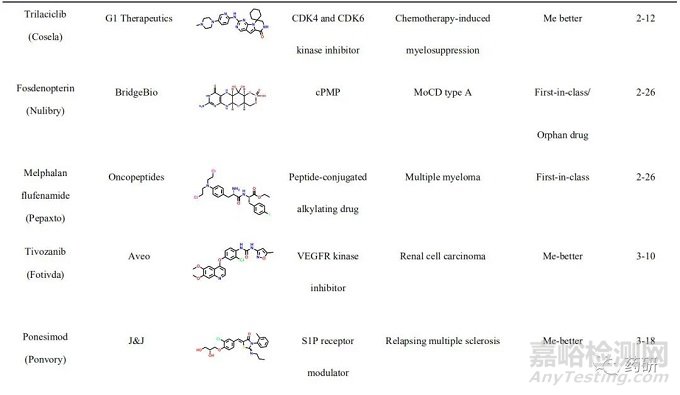
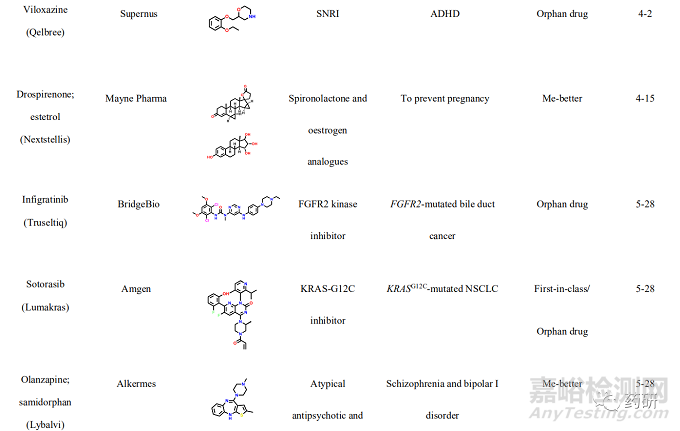
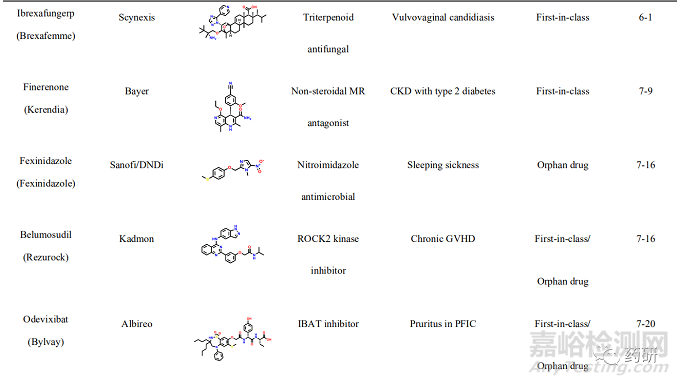
2021年, 美国FDA药物评价和研究中心(Center for Drug Evaluation and Research, CDER)共批准上市新药50个, 包括36个新分子实体和14个生物药物, 略低于2020 年(53个)。近五年的平均新药批准数量为51个, 远超于10年前24个的年均批准数量。从获批新药的适应症分布可以发现, 抗肿瘤药物获批15个(30%), 仍占主导地位;治疗自身免疫性疾病的相关药物获批7个(14%)、治疗内分泌代谢的相关药物获批6个(12%)、治疗神经相关疾病和遗传性疾病的药物各获批5个(10%)。值得一提的是, 2021年获批的新药分子中, 有34个新药获得了“优先评审”资格, 加速获批上市;26个新药获得了孤儿药上市资格, 使得以孤儿药资格获批的药物连续多年超过50%。在今年获批的50个新药中, 有27个属于first-in-class的首创性药物(占比54%), 已经成为新药获批的主力军。其中, 小分子首创性药物正在逐渐成为新药研发中最重要的组成部分, 占据每年首创性药物的大部分比例。今年, 27 个首创性药物中包括15个首创性小分子药物(如表1所示)。例如:首个新生儿Fc 受体(FcRn)拮抗剂艾加莫德(efgartigimod), 用于治疗乙酰胆碱受体阳性的重症肌无力患者;首个口服给药选择性补体C5a受体(C5aR)抑制剂阿伐可泮(avacopan), 用于辅助治疗两种主要的抗中性粒细胞胞浆自身抗体相关血管炎症;首个靶向KRAS G12C 突变蛋白的索托雷塞(sotorasib), 打破了KRAS 一直以来被认为是“不可成药”靶标的魔咒, 用于治疗KRAS G12C 突变、局部晚期或转移性 NSCLC 成人患者;首个靶向BCR-ABL1 蛋白肉豆蔻酰口袋的变构抑制剂阿思尼布(asciminib), 通过非ATP竞争的变构调控机制将BCR-ABL1锁定于非活性构象, 用于治疗先前已接受过至少2种酪氨酸激酶抑制剂(TKI)治疗、费城染色体阳性的慢性髓性白血病成人患者等;首个通过抑制HIF-2α治疗希佩尔·林道综合征相关的肾性癌症的小分子抑制剂贝组替凡(belzutifan)等。此外, 还有多个首创性的生物药物和诊断试剂, 包括首款阿尔茨海默病新疗法阿度卡单抗(aducanumab), 首个降低“坏胆固醇”的 siRNA 疗法英克司兰(inclisiran)等, 均通过全新的作用机制实现了治疗目的, 其中很多疾病是首次得到有效的治疗药物, 为后续的药物研发提供了广阔的思路与研究借鉴。


一、索托雷塞(sotorasib)——全球首个靶向KRAS G12C 突变蛋白的小分子抑制剂用于治疗KRAS G12C 突变的非小细胞肺癌
1.1 研发背景
RAS家族基因的发现由来已久, 是最早发现的致癌基因家族之一[6]。RAS 基因的突变会导致下游信号通路的永久性激活, 最终导致细胞的恶性增殖与分裂。RAS 家族基因主要包括NRAS、GRAS和KRAS三种类型, 尽管三种基因的突变均与癌症直接相关, 但KRAS的突变最为常见。现有研究表明大约在1/4 的人类肿瘤中都发现了KRAS的突变, 因此号称“癌症之王”。其中, KRAS G12C的位点突变在 NSCLC 患者中常见报道, 约占所有KRAS 突变的44%[7]。据统计, 大约有14%的 NSCLC 患者携带有KRAS G12C 突变基因, 全球每年超过10万人被确诊为KRAS G12C突变, 临床需求很大。在索托雷塞获批上市前, 对于接受一线治疗失败的 KRAS G12C 突变的NSCLC 患者无针对性的治疗方法, 缓解率低于 10%, 生存期仅为4个月。
和其他传统的蛋白靶标不同的是, KRAS蛋白结构复杂、动态性较高、底物GTP结合力强, 区域极性大, 缺乏明确且适宜的分子结合腔, 因此对小分子药物设计提出了极高的挑战。由于早期的研究案例多以失败告终, KRAS 在过去一直被认为是“不可成药”靶点, 其抑制剂的设计也是科学界公认的药物设计难题。索托雷塞的成功获批, 不仅为众多KRAS G12C 突变的NSCLC 患者带来福音, 同时也打破了KRAS不可成药的魔咒, 为KRAS 以及此类靶标的药物研发提供了重要的分子设计思路与开拓性的研究方法, 具有里程碑式的重要意义。
1.2 研发过程
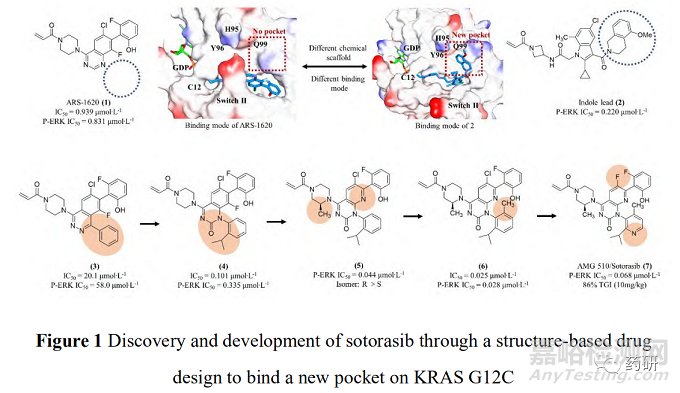
索托雷塞是由美国安进公司在大量的前期研究基础上通过合理药物设计得到的。在认识到靶向KRAS G12C的潜在治疗前景后, 研究人员开始尝试各种策略试图得到有效的小分子抑制剂。2013年, Shokat 等[8]在《Nature》上首次报道了利用共价结合策略合理设计小分子结合于KRAS 的C12 半胱氨酸残基上, 实现了对KRAS G12C 突变的肿瘤细胞增殖的有效抑制。在经过几轮结构优化后得到了活性和理化性质较优的苗头分子ARS-1620(1)可供后续的体内实验研究。共晶复合物结构表明, ARS-1620(1)的结合位点与GTP不同, 可在GDP结合的状态下与 KRAS G12C的变构位点共价结合(图 1)[9]。
研究早期, 安进公司的科学家也筛选了大量的含有半胱氨酸反应片段的小分子库, 得到了一系列具有潜在活性的小分子化合物。经多轮的结构优化后最终得到了具有吲哚骨架的先导化合物2, 表现出了较好的抑制活性。有趣的是, 化合物2的共晶复合物结果表明, 虽然共价结合位点表现出与ARS-1620(1)相似的结合模式, 但在由 H95、Y96 和Q99组成的浅表位点的结合模式却完全不同。ARS-1620(1)的分子片段没有伸向此结合腔, 而吲哚骨架的先导物2却可占据此位点。由于吲哚骨架的先导物2清除率高、口服生物利用度低, 无法进行体内实验, 故考虑将吲哚骨架先导物2的优势片段(即可占据浅表位点的结构片段)与ARS1620(1)进行分子融合, 以期获得具有更优成药性的小分子抑制剂[10]。
将ARS-1620(1)与吲哚骨架先导物的骨架叠合发现, ARS-1620(1)的嘧啶环一侧可以继续延伸刚性片段, 模拟吲哚先导物2的优势骨架, 得到了化合物3, 其对 KRAS G12C的抑制剂活性和下游P-ERK的抑制活性均有所下降(IC50 = 20.1 μmol∙L-1, P-ERK IC50 = 58.0 μmol∙L-1)。随后, 进一步探索新延伸出的苯环片段的构效关系发现在苯环2位引入异丙基并将哒嗪母核替换为嘧啶酮后的化合物4可使活性大幅提高(IC50 = 0.101 μmol∙L-1, P-ERK IC50 = 0.335 μmol∙L-1)。共晶复合物结果也表明, 新延伸出的分子片段可以有效占据由H95、Y96 和Q99组成的浅表新腔穴, 抑制活性已经超过模板分子ARS-1620(1)。后续针对哌嗪环和苯并嘧啶的构效关系研究得到的化合物5活性进一步提升(P-ERK IC50 = 0.044 μmol∙L-1), 由于5存在手性位点, 研究表明只有R 构型可以有效匹配KRAS蛋白空腔, S构型活性大幅下降。这一手性构型问题也提示需要考虑延伸出的苯环的构象扭转问题, 因此尝试在新延伸出的苯环上添加小基团取代基, 试图通过分子内位阻限制苯环的构象扭转。这一策略得到了成功, 结果表明化合物6的活性进一步提升(IC50 = 0.025 μmol∙L-1, P-ERK IC50 = 0.028 μmol∙L-1), 并表现出一定的口服生物利用度(F = 21%)。最后一步的结构优化策略聚焦于提升化合物的水溶性和提高极性表面积, 最终获得了AMG 510 (7), 即索托雷塞, 呈现最优的活性与成药性质, 体内抑瘤率(TGI%)高达 86% (图 1)[10]。
1.3 治疗应用
索托雷塞是全球第一个获批的KRAS G12C 小分子抑制剂, 通过共价结合KRAS G12C 位点抑制突变蛋白的功能, 进而干扰RAS-RAF-MEK-ERK信号通路, 在KRAS G12C 突变的晚期非小细胞肺癌患者上表现出优异的疗效。值得一提的是, 索托雷塞开展临床试验的患者均为接受过不止一种化疗或免疫疗法后病情继续恶化, 非常难治的晚期患者。临床结果表明, 索托雷塞的客观缓解率(Objective Response Rate, ORR)为37.1%, 疾病控制率为80.6%。中位反应持续时间为10 个月。81%的患者都出现了肿瘤缩小的情况, 肿瘤缩小的平均值为60%。与此同时, 索托雷塞也表现出了良好的耐受性, 不良反应为食欲下降、腹泻、乏力、头痛、咳嗽、潮热和恶心, 并且没有发现剂量限制毒性和与药物有关的4级以上不良反应[10-14]。索托雷塞的成功不仅仅是一个药物的成功, 也是药物设计的巨大进步。索托雷塞不仅造福了数万KRAS基因突变的癌症患者, 也为更多的突变靶标的药物设计提供了宝贵的研究思路与方法借鉴。
二、阿思尼布(asciminib)——全球首个 STAMP 变构抑制剂用于治疗慢性髓系白血病
2.1 研发背景
慢性髓系白血病(Chronic Myeloid Leukemia, CML)为严重威胁人类健康的恶性肿瘤疾病, 其发病过程主要由BCR-ABL1 融合蛋白而驱动[15]。早在 2001 年, 伊马替尼(imatinib)就作为第一个靶向ABL1 ATP位点的激酶抑制剂上市, 开启了激酶类药物研究的热潮, 使得CML的生存期大大延长, 同时也创造了极高的经济价值。在阿思尼布上市前, 所有成功上市的激酶类药物的作用机制与伊马替尼类似, 均为靶向ATP结合位点的竞争型抑制剂, 并逐渐成为临床用药的首选, 有效延长了患者的生存期。然而, 肿瘤细胞是个“狡猾的对手”, 患者长时间用药后, 药物靶向激酶的ATP位点常见各种耐药突变, 超过50%的患者在使用激酶类抑制剂后会产生不同程度的耐药, 目前尚未有较好的解决方案[16]。
对于BCR ABL1 而言, T315I 突变最为常见。CML患者通过第一代抑制剂伊马替尼治疗后的10年生存率可达85%~90%, 但在长时间用药过程中发现了包括T315I 在内的多种突变, 使得伊马替尼的疗效大幅下降[17]。以达沙替尼(dasatinib)和尼洛替尼(nilotinib)为代表的第二代抑制剂虽对多种ABL1 突变有效, 但仍未解决T315I 突变的问题。第三代ABL1 抑制剂泊那替尼(ponatinib)虽然可一定程度上逆转 T315I 耐药, 但不良反应严重, 使用受限。由此可见, 通过分子结构的迭代不能解决ABL1 的T315I 耐药问题, 需要从药物设计的源头进行考虑。
2.2 研发过程
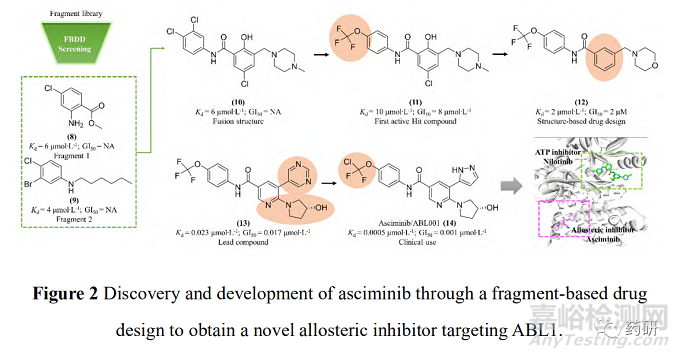
诺华公司为解决ABL1 蛋白的T315I 突变问题, 设计了非ATP 竞争性的变构调控策略(图 2)。为此, 首先通过基于碎片的药物设计进行片段筛选。实验中发现了超过30个片段对ABL1 蛋白有一定强度的亲和力, 其中化合物片段8的 Kd 值为6 μmol∙L-1, 作为一个分子量仅有185.6 的分子片段, 已有较高的配体利用效率(LE = 0.6), 具有较大的优化空间。但随后的实验表明化合物8并不能抑制 ABL1的ATP 酶活, 也没有细胞活性, 这表明此分子可能不结合于ATP 位点。随后的共晶复合物结构证明了这一结果, 揭示了一个可结合于肉豆蔻酰腔穴的全新位点。为了进一步提升片段分子8的活性, 去除芳香环上的甲酯并引入脂肪碳链得到化合物 9, 其Kd值为 4 μmol∙L-1, 但仍无细胞活性。通过融合8和9的结构片段, 得到了化合物10(Kd =6 μmol∙L-1), 这成为后续分子优化的骨架结构。至此, 研究人员已经意识到了此类分子是结合于全新的变构结合腔而表现出一定的亲和力, 但如何进行后续的分子设计诱导 ABL1 转化为非活性构象成为了难点问题[18]。
为了解决这一问题, 深入探索ABL1活性构象与非活性构象的转变关系, 使 用了 N15 标记的二维核磁实验进行蛋白构象的探索。基于前期的经验, 在化合物10 的结构骨架上引入CF3O-基团得到的化合物11产生了抑制活性(Kd = 10 μmol∙L-1; GI50 = 8 μmol∙L-1), 表明CF3O-基团是诱导非活性构象产生的重要基团, 化合物11也成为了苗头化合物。通过基于结构的药物设计开展进一步的优化得到了活性提升的化合物12(Kd = 2 μmol∙L-1; GI50 = 2 μmol∙L-1)。通过分析化合物的结合模式, 发现12中的吗啉环电子云密度无序, 与结合作用力小, 因此试图通过引入芳香环提高疏水作用力。这一想法在化合物13上得到了验证, 通过引入嘧啶环和 3-羟基吡咯烷基使得化合物13的活性大幅提升(Kd = 0.023 μmol∙L-1; GI50 = 0.017 μmol∙L-1), 随后被视为先导化合物进行后续的成药性优化。最终, 用氯原子取代CF3O-基团中的一个氟原子实现了活性的进一步提升, 使用吡唑环代替嘧啶环降低了 hERG 毒性, 得到的ABL001(14)即为阿思尼布[19]。阿思尼布的成功, 不仅提供了靶向ABL1 的全新思路, 也明确了ABL1 活性构象与非活性构象的转变过程, 结构优化巧妙, 成为变构抑制剂研究的典范。
2.3 治疗应用
尽管在过去几十年中, 慢性髓性白血病(CML)的治疗已经取得巨大的成功, 多数患者生存期可延长至10年以上, 但药物均为激酶的ATP抑制剂, 存在耐药性和位点突变的风险。阿思尼布不仅对野生型的ABL1激酶有抑制活性, 得益于独特的变构调控机制, 对T315I 突变的ABL1同样具有高活性, 也大大降低了 ATP 类抑制剂潜在的脱靶效应。目前, 阿思尼布可用于治疗先前已接受过至少 2 种酪氨酸激酶抑制剂(Tyrosine Kinase Inhibitor, TKI)治疗、费城染色体阳性慢性髓性白血病慢性期(Ph+CML-CP)成人患者以及携带T315I 突变的Ph+CML-CP 成人患者。
三、贝组替凡(belzutifan)——全球首个 HIF2α 抑制剂用于治疗希佩尔·林道综合征相关癌症
3.1 研发背景
希佩尔·林道综合征(Von Hippel-Lindau, VHL)是一种罕见的遗传性疾病, 目前全球患者超过20万人, 在贝组替凡(belzutifan)获批前无系统的治疗方案。不幸的是, VHL 患者一生有超过70%的概率发展成为肾细胞癌(Renal Cell Carcinoma, RCC)或其他相关癌症, 成为VHL患者主要的死亡原因。RCC是最常见的肾癌类型, 约占总量的90%, 死亡率接近50%[20]。VHL 患者中的抑癌基因VHL会发生突变(概率超过90%), 导致其活性异常并激活肿瘤患者体内的缺氧诱导因子-2α(HIF-2α)蛋白[21]。HIF-2α 的激活与聚积会向人体错误地发出缺氧信号, 进而导致下游通路的异常并激活血管生成, 从而促进肿瘤的增殖与生长[22]。因此, 抑制HIF-2α 成为一种潜在的肾癌治疗方案。
3.2 研发过程
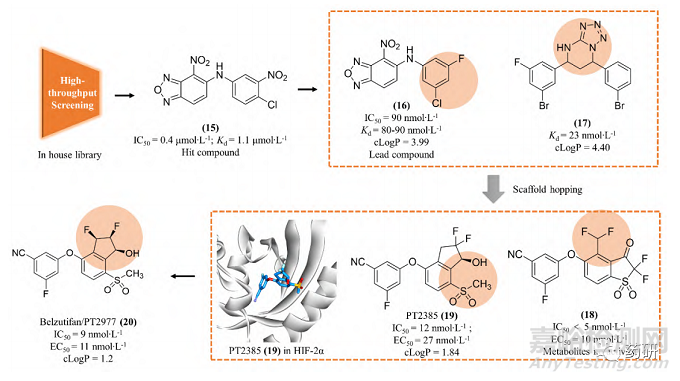
为了获得有效的HIF-2α 蛋白小分子抑制剂, Bruick 和 Tambar 等[23]在2012 年通过基于荧光接近的AlphaScreen方法筛选了包含超过20 万个的小分子化合物库, 得到了20 个具有潜在活性的分子骨架。其中, 化合物15 可以通过结合至 HIF-2α PAS-B 内部空腔而表现出一定的抑制活性(IC50 = 0.4 μmol∙L-1; Kd = 1.1 μmol∙L-1), 被视作苗头化合物。分析15 的分子结构, 包含3个组成部分, 即两侧的芳香环系和中间的连接基。随后对这3个部分开展了深入的构效关系研究, 得到的化合物16 抑制活性和亲和力均显著提高(IC50 = 90 nmol∙L-1; Kd= 80~90 nmol∙L-1)。用高活性的16开展了初步的机制探索, 表明16可以诱导HIF-2α PAS-B 的空间构象变化, 并显著阻断HIF-2的二聚化过程, 同时对 HIF-1α几乎没有亲和力, 选择性高。同时还发现了具有高亲和力骨架结构的化合物17(Kd = 23 nmol∙L-1)[24]。
Peloton Therapeutics 公司基于化合物16 和17的分子结构开展了深入的成药性优化研究。通过融合其优势片段, 作骨架迁越(scaffold hopping)等方法, 最终得了两类具有高活性的HIF-2α 抑制剂, 即化合物18(IC50 < 5 nmol∙L-1; EC50 = 10 nmol∙L-1)和19(IC50 = 12 nmol∙L-1; EC50 = 27 nmol∙L-1)。体外活性结果显示18 的活性高于19, 然而体内研究发现18易被氧化代谢为无活性的代谢物, 体内活性差、口服生物利用度低。因此, 化合物19(PT2385)被视为最早的HIF-2α小分子抑制剂而进入临床研究[25]。然而, 临床实验中发现化合物19对ccRCC的疗效不佳, 一度在业内引起HIF-2α 靶标是否有效的争论。Peloton Therapeutics 公司并未放弃, 在2019年报道了化合物20(PT2977)作为改进后的HIF-2α小分子抑制剂(IC50 = 9 nmol∙L-1; EC50 = 11 nmol∙L-1), 同时阐明了早期抑制剂失效的原因:化合物19环戊烷上的羟基极易被葡萄糖醛酸化, 生成的代谢物(conjugate)活性大幅下降, 导致体内暴露量降低而疗效差。为解决这一问题, 通过改变环戊烷上氟原子的位置, 以期降低邻位羟基的亲核性并降低葡萄糖醛酸化[26]。最终得到的化合物20[贝组替凡(belzutifan)]不仅活性提高, PK 性质也实现飞跃, 在临床试验中表现出理想的疗效, 最终成功上市(图 3)。

3.3 治疗应用
贝组替凡(belzutifan)是FDA 批准的首个HIF-2α 抑制剂疗法。通过抑制HIF-2α, 贝组替凡(belzutifan)可降低与细胞增殖、血管生成和肿瘤生长相关的HIF-2α 靶基因的转录和表达。在VHL相关的RCC患者中的客观缓解率达到 49%, 其中有超过一半的患者疗效持续时间超过12个月, 具有令人满意的有效性。治疗常见的不良反应包括:血红蛋白减少、贫血、疲劳、肌酐增高、头痛、晕眩、葡萄糖水平升高、恶心等。发现HIF-2α 的VHL生物学研究获得了2019 年的诺贝尔生理学或医学奖, 贝组替凡(belzutifan)的成功又为靶向 HIF-2α 的研究锦上添花, 也为未来更多缺氧因子通路的相关药物研究提供了宝贵思路。
四、 结语与展望
除了以上浅析的首创性药物之外, 本年度获批的新药分子涉及至少7个全新的药物靶标, 包括首个新生儿Fc 受体(FcRn)拮抗剂艾加莫德(efgartigimod), 用于治疗抗乙酰胆碱受体抗体阳性(AChR Ab+)的全身重症肌无力成人患者;首个抗胸腺间质淋巴细胞生成素单克隆抗体特泽芦人单抗(tezepelumab), 用于治疗年龄≥12 岁的严重哮喘儿科患者和成人患者;首个口服给药选择性补体C5a受体(C5aR)抑制剂阿伐可泮(avacopan), 用于辅助治疗两种主要的抗中性粒细胞胞浆自身抗体(ANCA)相关血管炎:显微镜下多血管炎(MPA)和肉芽肿伴多血管炎(GPA);首个I 型干扰素受体(IFNAR)抗体阿伏芦人单抗(anifrolumab), 用于治疗正在接受标准治疗的中度至重度系统性红斑狼疮(SLE)成人患者;首个血管生成素样蛋白3(ANGPTL3)抑制剂依维库人单抗(evinacumab), 用于其他降脂疗法的辅助疗法, 治疗年龄≥12 岁纯合子家族性高胆固醇血症(HoFH)儿童和成人患者等等。
2021年, 我国药品监督管理局(NMPA)共批准了 76 款新药, 其中国产创新药 27 款, 中药创新药12款, 进口新药37款, 获批数量创下新高。其中包括多个我国自主研发的小分子药物, 例如:恒瑞医药研发的首款国产原创SGLT2 抑制剂恒格列净(henagliflozin)和仅用8个月就通过优先评审获批上市的CDK4/6 抑制剂达尔西利(dalpiciclib)(国际上此靶点第五款新药);亚盛医药研发的中国首个第三代 BCR-ABL抑制剂奥雷巴替尼(olverembatinib);微芯药业研发的全球首个PPAR 全激动剂西格列他钠(chiglitazar sodium);和记黄埔医药研发的国内首个高选择性MET抑制剂赛沃替尼(savolitinib);百济神州研发的首款用于治疗铂敏感及铂耐药复发性卵巢癌的PARP抑制剂帕米帕利(pamiparib)等等。随着我国科研实力的不断提高, 相信在不久的将来, 国内的医药企业和研究院所可以实现更多原创性的突破, 让更多的首创性药物分子贴上“中国制造”的标签。

来源:Internet


