医疗器械作为一种强监管产品,整个生命周期包括研发、生产、审批、运输、存储、销售、使用等一系列过程,医疗器械各个环节都需要科学管理,否则可能发生医疗损害责任纠纷或医疗事故,对患者生命安全和健康都将造成重大威胁。医疗器械研发包括产品研制、临床评价、注册许可等流程。对于不同类别的医疗器械,审批和监管要求也各不相同。
医疗器械分类
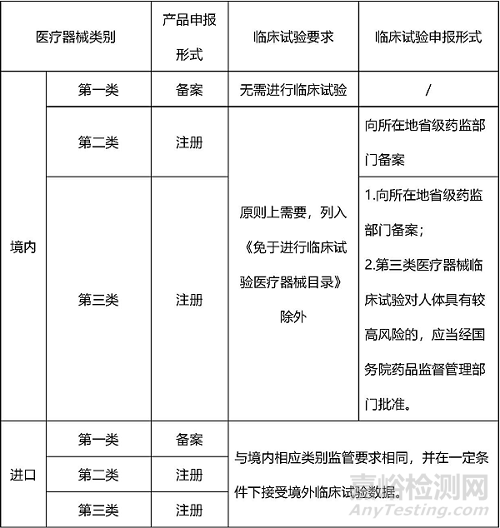
国家对医疗器械按照风险程度实行分类管理。医疗器械从风险程度高低分为三类,并采用不同的管理方式:第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械,如手术刀、止血钳、压舌板等。第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械,如面部提拉线、面部埋植线等。第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械,如射频治疗仪、超声治疗仪、水光针等。根据《医疗器械分类规则》规定,评价医疗器械风险程度,应当根据医疗器械的预期目的、结构特征、使用形式、使用状态等因素综合判定。
医疗器械研发及监管
医疗器械研发环节一般包含从产品研制到获得注册许可或完成备案的流程,具体包含产品研制、注册检验、临床评价、注册申请、技术审评、注册许可等流程。医疗器械的临床评价和注册许可是医疗器械研发阶段的监管重点。
医疗器械临床评价是指申请人采用合理的方法对临床数据进行分析,以确认产品是否满足使用要求而进行确认的过程,目前临床评价主要有以下三种方式:
一是直接豁免:列入《免于进行临床试验的医疗器械目录》产品的临床评价。
二是同品种对比:通过对同品种医疗器械临床文献资料、临床数据进行分析评价,证明医疗器械安全、有效。
三是临床试验:按照国家药品监督管理部门的规定,进行医疗器械临床评价时,已有的临床文献资料和临床数据不足以确认产品安全、有效的医疗器械,应当开展临床试验。
医疗器械的监管方式主要有以下几种:
图片医疗器械研发合规风险总结
1. 医疗器械临床试验
为加强医疗器械临床试验的监督管理,国家食品药品监管总局每年都会对医疗器械临床试验进行监督抽查并发布《总局关于开展医疗器械临床试验监督抽查工作的通告》,对临床试验数据的真实性和合规性进行检查,未发现真实性和合规性问题的,视为临床试验符合要求。首先,临床试验存在真实性问题情况有以下情形:一是注册申请提交的临床试验资料与临床试验机构保存的相应临床试验资料不一致的;二是临床试验数据不能溯源的;三是受试产品/试验用体外诊断试剂或试验用样本不真实的。其次,未发现真实性问题,但临床试验过程不符合医疗器械相关规定要求的,判定为存在合规性问题。再次,未发现真实性和合规性问题的,判定为符合要求。对存在真实性问题的,可能会作出责令改正、停止临床试验或进行罚款等处罚。对仅存在合规性问题的,综合评价注册申请资料和监督检查发现的问题,作出是否批准注册的决定。
2. 医疗器械注册备案
为确保医疗器械顺利通过审批,申请人或备案人可能会出现提交虚假注册资料或备案资料、隐瞒真实情况或提供虚假信息等情况,《医疗器械注册与备案管理办法》均对上述情况进行了规定,在审评、核查、审批过程中发现涉嫌存在隐瞒真实情况或者提供虚假信息等违法行为的,申请人不得撤回医疗器械注册申请。对于已受理的注册申请,有证据表明注册申请资料可能虚假的,药品监督管理部门可以中止审评审批。经核实后,根据核实结论继续审查或者作出不予注册的决定。对于拟上市的医疗器械无法证明产品安全有效、质量可控等情况的,药品监督管理部门作出不予注册的决定。
3. 医疗器械专利
医疗器械专利研发完成以后,更新迭代进程较慢,而专利权具有独占性、排他性等特点,医疗器械研发过程容易产生专利侵权纠纷。从医疗器械研发的外部风险来说,如果专利技术方案落入他人的专利权保护范围,可能被专利行政部门予以驳回;即使被授予专利,但权利人认为该专利权的授予不符合专利法规定的,可以请求专利复审委员会宣告该专利权无效。从医疗器械的内部风险来说,即在单位未与单位内部发明人、设计人明确职务发明的权利归属的情况下,员工对职务发明的权属提出异议或者企业对发明人、设计人给予职务发明奖励和报酬而引发相应民事争议的法律风险。