摘 要 / Abstract
习近平总书记强调,生物医药产业是关系国计民生和国家安全的战略性新兴产业。要加强基础研究和科技创新能力建设,把生物医药产业发展的命脉牢牢掌握在我们自己手中。医疗器械产业作为生物医药产业的重要组成部分,医疗器械注册审查指导原则的科学性是其基础研究能力的重点体现之一,医疗器械上市前安全有效性评价的科学性是其科技创新能力的影响因素之一。本文从指导原则体系的规划、程序、分布、结构、管理等方面,对比分析中国、美国、欧盟、日本等指导原则现状,呈现我国指导原则体系建设的特点。按照国家药监局“提前介入、一企一策、全程指导、研审联动”的要求,通过调研明确我国指导原则体系建设的下一步重点工作,以期进一步发挥指导原则提高科学审评能力、促进产业高质量发展的作用。
General Secretary Xi Jinping has stressed that the biomedical industry is a strategic emerging industry crucial to national economy, people's livelihood, and national security. It is necessary to strengthen basic research and scientific and technological innovation capabilities and maintain control over the development of the biomedical industry. As an important part of the biomedical industry, the quality of guidance practices for medical device registration review is a key manifestation of its basic research ability, and the scientific evaluation of safety and effectiveness of medical devices before premarket is a determinant of its scientific and technological innovation ability. This paper compares and analyzes the current status of guidance practices in China, the United States, the European Union, and Japan from six aspects: planning, procedures, distribution, structure, and management of guidance practices systems, highlighting the characteristics of China’s Good Guidance Practices system construction. According to NMPA’s requirements of "early intervention, tailored approach for each enterprise, whole-process guidance, and integration of research and evaluation", the next key work for the construction of Good Guidance Practices in China is clarified through investigation, aiming to maximize the role of Good Guidance Practices in enhancing scientific evaluation capabilities and promoting high-quality development of the industry.
关 键 词 / Key words
医疗器械;指导原则;体系;审评科学;高质量发展
medical devices, guidance practices, system, evaluation science, high-quality development
当前,全球医疗器械科技创新方兴未艾,市场规模持续扩大。弗若斯特沙利文机构(Frost & Sullivan)预测,未来5 年医疗器械产值年均复合增长率将达4.4%,到2027 年,医疗器械产业规模将突破5100 亿美元[1]。医疗器械涉及声、光、电、磁、材料、临床医学等多个专业,具有学科交叉广泛、技术知识密集、产业创新活跃等鲜明特点,是衡量一个国家/ 地区高科技发展综合实力的重要标志之一[2]。
习近平总书记强调,生物医药产业是关系国计民生和国家安全的战略性新兴产业,要加强基础研究和科技创新能力建设,把生物医药产业发展的命脉牢牢掌握在我们自己手中[3]。医疗器械产业是生物医药产业的重要组成部分之一,与药品、医疗技术并驾齐驱,共同构成完整的医疗服务体系[4]。医疗器械监督管理遵循风险管理、全程管控、科学监管、社会共治的原则[5]。对医疗器械进行上市前安全性、有效性的评价(技术审评)是国际通用的监督管理模式,具有高度的专业性和技术性。
医疗器械注册审查指导原则的科学性是医疗器械产业基础研究能力的重点体现之一,医疗器械上市前安全有效性评价的科学性是医疗器械产业科技创新能力的影响因素之一。医疗器械技术审评机构要加强基础研究和科技创新能力建设,构建与我国医疗器械产业发展相适应的指导原则体系,筑牢审评科学的技术文件根基,从而推动医疗器械监管效能,进一步助力、引导医疗器械产业高质量发展[6]。
1、指导原则体系
医疗器械注册审查指导原则(以下简称指导原则)是由医疗器械技术审评机构制定,面向技术审评人员、注册申请人、社会公众,用以配套上市前监管政策的执行或明确注册技术审评要求的指导性文件。指导原则体系是医疗器械技术审评机构制定、发布和使用指导原则的程序和规范[7]。
从内容来看,指导原则可分为通用类指导原则和产品类指导原则。通用类指导原则是以《医疗器械监督管理条例》以及配套法规、规范性文件为基准,进一步明确注册申报的程序要求、资料内容、评价要点等的文件。产品类指导原则是以《医疗器械安全和性能的基本原则》为核心,基于产品特点,明确监管产品的设计、生产、验证、确认、标识等内容的文件。
指导原则是医疗器械技术审评过程中制定的共识文件,同时具有解读法律法规规范、指导注册申报和适应产业发展的属性。指导原则作为医疗器械技术审评的新工具、新方法、新标准,是实现审评科学的必经路径[8],是医疗器械监管科学行动计划的重要产出,是医疗器械产业高质量发展的可靠保证。同时,考虑到医疗器械结构组成复杂,风险程度不一,从事审评的工作人员,因知识结构和专业背景各不相同,对产品风险和受益的分析往往具有一定的主观性,指导原则体系建设的作用在于审评思维的培养和审评认知的统一,使得审评员及时跟踪产品相关的基础研究成果,熟悉掌握法规要求和技术知识,提炼审评获得经验,梳理转化成为指导原则,为审评提供技术支持[4]。
2、指导原则体系的国际比较
使用指导原则开展医疗器械上市前安全有效性评价是许多国家施行的监管模式。不同监管机构制定的指导原则基于自身监管实际,在指导原则的制定主体、编制目的、管理模式等方面存在异同,详见表1。
(一)中国指导原则体系概况
中国第三类指导原则由国家药品监督管理局医疗器械技术审评中心(以下简称器审中心)制定并发布。第二类指导原则由器审中心组织省级医疗器械审评机构编制,器审中心负责相关组织协调、质量把控和发布。
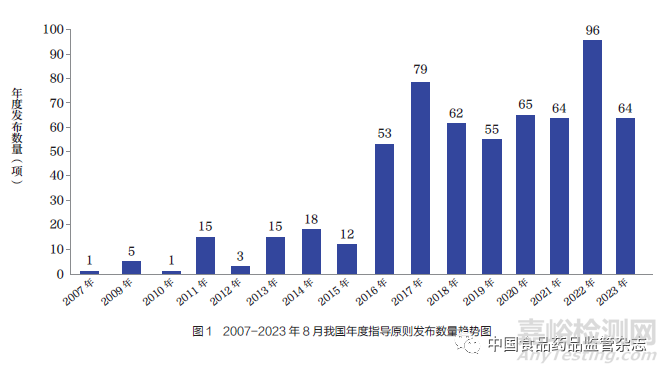
2007 年,我国发布第一项医疗器械指导原则。截至2023 年8 月底,我国医疗器械指导原则制修订数量已达608 项。自2016 年“十三五”规划以来,发布数量显著提升,“十三五”“十四五”规划期间发布的指导原则数量(含修订)为538 项,占发布总数的88.5%。2022 年发布了96 项指导原则,为历年发布数量之最(图1)。
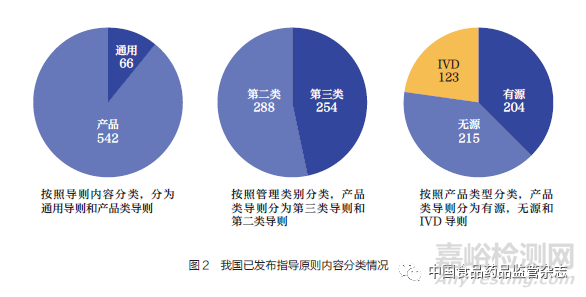
我国制修订的通用类指导原则为66 项,产品类指导原则为542 项。产品类指导原则按照管理类别包括第三类指导原则和第二类指导原则。产品类指导原则按照产品类型包括有源类、无源类和体外诊断试剂类产品指导原则(图2)。
(二)美国指导原则体系概况
美国医疗器械监管起步早,经过长期的经验积累,美国食品药品监督管理局(Food and Drug Administration,FDA)指导原则体系建设较为成熟。美国有关医疗器械指导原则的制定等工作由FDA 下属医疗器械和辐射健康中心(Center for Devices and Radiological Health,CDRH)承担。FDA 于1997 年发布了良好指导原则规范(Good Guidance Practices,GGP)草案,明确了FDA 指导原则文件制定、发布和使用的基本要求[7]。该文件申明了FDA 指导原则不具有强制力,但同时强调了对FDA 工作人员的约束力,FDA 工作人员仅在有正当理由和监管同意的情况下才可违背指导原则文件要求开展审评工作。
CDRH 公开了指导原则制定标准操作规程,详细介绍了指导原则发文流程。此外,CDRH 每财年发布指导原则清单,包括本年度拟制定指导原则和拟开展回顾性审查的指导原则,对部分既往已发布指导原则进行回顾性审查,接收外部反馈信息,并适时进行修订,进一步加强了监管的透明度和公众参与性。截至2023 年8 月底,通过FDA 官方网站查询到,FDA 医疗器械指导原则文件共约1100 余项,其中通用程序性指导原则84 项,通用技术性指导原则430 项,有源产品类指导原则284 项,无源产品类指导原则182 项,体外诊断试剂(In Vitro Diagnostic Devices,IVD)产品类指导原则128 项。
(三)欧盟指导原则体系概况
欧盟由第三方公告机构(Notified Body,NB)进行上市前审评,欧盟委员会及成员国家主管部门负责监督和管理第三方公告机构。为帮助医疗器械生产企业、公告机构等利益相关方更好地理解、执行医疗器械相关法规,欧盟委员会发布了一系列指导原则文件。
欧盟医疗器械指导原则文件类型包括:对特定医疗器械产品定义范围的进一步阐释,医疗器械产品性能评估基本要求,医疗器械分类规则,公告机构审查认证评定程序,临床评价相关要求等。
自医疗器械法规(第 2017/745 号)和体外诊断医疗器械法规( 第 2017/746 号) 实施以来,欧盟委员会下设的公共卫生部的医疗器械协调小组(Medical Device Coordination Group,MDCG),根据欧盟委员会的要求,组织制定了多项阐释医疗器械法规的指导原则[9],加大了指导原则的编制力度,进一步规范了公告机构的审查要求。据不完全统计,欧盟委员会已发布指导原则150余项,征求意见稿60 余项。
(四)日本指导原则体系概况
日本厚生劳动省(Ministry of Health, Labour and Welfare,MHLW) 是日本负责医疗卫生和社会保障的主要部门,其下设独立法人机构日本药品和医疗器械管理局(Pharmaceuticals and Medical Devices Agency,PMDA) 是医疗器械监管的主要职责部门。日本医疗器械的上市前许可方式为委托管理与直接管理相结合,对于有且符合该产品适用的审查依据性文件的医疗器械,应当采取第三方认证机构认证方式获得上市许可,否则需PMDA 进行审评、日本厚生劳动省承认后,获得上市许可。
为适应不同的上市前许可路径,日本医疗器械注册审查依据性文件也存在多种类型,包括认证基准、承认基准以及审查指导原则[10]。认证基准由厚生劳动省发布,用于供审查医疗器械申请的注册认证机构评估医疗器械的合规性。承认基准是指厚生劳动省认可的国际标准化组织(International Organization for Standardization,ISO)或国际电工委员会(International Electrotechnical Commission ,IEC) 等国际组织发布的文件。审查指导原则同样由厚生劳动省发布,主要内容为描述评估医疗器械安全性和有效性所需的主要技术要求和(或)验收标准等,帮助医疗器械生产企业更有效地对医疗器械进行PMDA 审查。截至2023 年8 月底,日本厚生劳动省制定了950 个认证基准、44 个承认基准和10 个审查指导原则。
3、我国指导原则体系的建设特点
通过对比分析中国、美国、欧盟、日本等国际监管机构指导原则体系概况,我国指导原则体系呈现出全面(基本覆盖全部子目录)、完善(基本建立指导原则体系)、丰富(指导原则体系有效延伸)、开放(竞速真实世界研究赛道)的特点。
(一)基本覆盖全部子目录
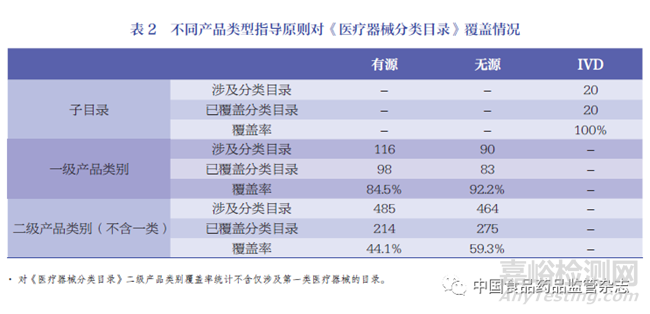
为分析我国医疗器械指导原则结构的完整性,审评人员曾在不同时期统计过产品类指导原则对应一级产品类别的覆盖率[11-12]。目前,医疗器械产品类指导原则已全部覆盖《医疗器械分类目录》的22 个子目录,覆盖率100% ;覆盖一级产品类别181 个,覆盖率为87.9% ;覆盖二级产品类别489 个,覆盖率为51.5%(不含第一类医疗器械)。已发布指导原则对医疗器械分类目录按产品类别有源、无源、IVD 分类覆盖情况见表2。《体外诊断试剂分类子目录》的20 个产品类别已实现全覆盖。
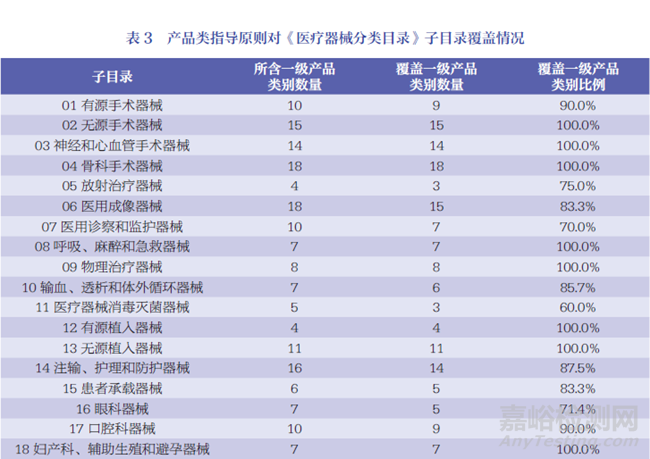
《医疗器械分类目录》22 个子目录中02、03、04、08、09、12、13、18、21 等9 项子目录的一级产品类别目录已实现指导原则的全覆盖,详见表3。
(二)基本建立指导原则体系
“十三五”以来,我国医疗器械指导原则编制工作成效明显,指导原则的质量和数量均有明显提升。为应对器械注册管理及审评工作开展的新形势,器审中心发布《医疗器械注册审查指导原则“十四五”规划》,对指导原则的编制工作进行科学规划,制定到2025 年的指导原则发展目标。建设指导原则体系过程中,器审中心坚持科学性原则,优化指导原则编制流程,编制了一系列制修订操作规范,加强过程管理,明确各环节的责任、时限要求。
在指导原则体系建立过程中,器审中心根据产品风险等级委托省级医疗器械审评机构编制第二类医疗器械指导原则,同时加强国家级和省级、省级技术审评机构之间的交流和共享,协同促进全国技术审评机构监管能力提升。鼓励生产企业、高等院校、科研机构、临床机构等社会力量广泛参与指导原则编制工作。通过器审云课堂的形式,组织编制人员录制指导原则培训视频。截至目前,器审中心已发布189 个第三类指导原则和42 个第二类指导原则的讲解视频。
(三)指导原则体系有效延伸
指导原则体系有效延伸至医疗器械技术审评要点(以下简称审评要点)的管理。技术审评机构以制定审评要点的形式,对注册申请项目相关技术信息和(或)相关领域科学研究的综合分析,提炼出对医疗器械安全性、有效性和质量控制方面有重要影响的技术要求或审评共识,用于指导和规范技术审评工作。随着同类产品的后续申报(一般为3个及以上),产品类审评要点会通过分技术委员会集体决策的形式进行完善,转化为产品类指导原则向社会发布。
器审中心对每一类医疗器械均编制了审评要点,并及时公开了一批创新医疗器械、临床急需医疗器械、应对公共卫生事件急需医疗器械等相关审评要点,缓解了指导原则从立项到发布周期较长无法匹配器械领域新技术、新材料更迭速度快的缺陷,发挥了指导原则体系及时规范、早期指导的作用。
截至2023 年8 月底,器审中心已制修订审评要点761 项,其中产品类审评要点333 项,占比 43.8%,临床评价类审评要点428 项,占比56.2%。
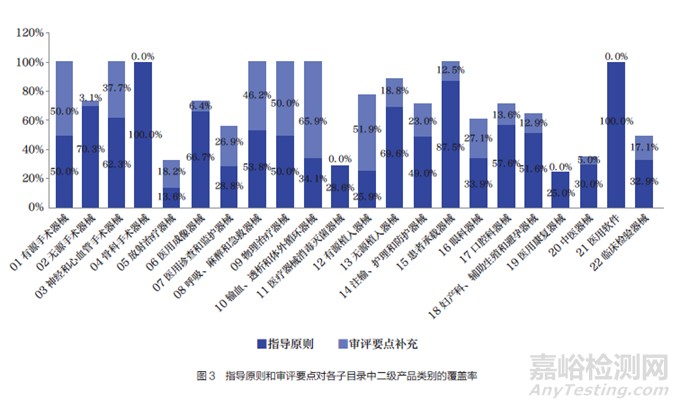
图3 为指导原则和审评要点在各子目录中二级产品类别的覆盖率,其中,深色为指导原则在子目录中二级产品类别的覆盖率,浅色为审评要点补充后在该子目录中二级产品类别的覆盖率。
(四)竞速真实世界研究赛道
2023 年8 月 30 日,美国 FDA 发布了《使用真实世界数据(Real World Data,RWD)和真实世界证据(Real World Evidence,RWE)支持药品和生物制品监管决策的考量因素》[13]的指导原则, 是FDA 建立的RWE 计划框架[14] 的最新文件,重申了真实世界研究(Real World Study, RWS) 无需申请新药临床研究(Investigational New Drug,IND),增加了受理前的沟通交流,明确了监管决策所期望的RWD/RWE。
RWD/RWE 在医疗器械领域同样有广阔的应用前景。RWD/RWE 用于临床评价,与传统临床试验形成互补,共同促进医疗器械监管和决策。中国药品监管科学行动计划将“真实世界数据用于医疗器械临床评价的方法学研究”列为第一批9 个行动计划项目中的一项,随后依然将“真实世界数据支持创新和临床急需医疗器械评价方法研究” 列为第二批10 个行动计划项目的一项。
依托于中国药品监管科学行动计划,RWS 取得一系列重要成果,包括发布《真实世界数据用于医疗器械临床评价技术指导原则(试行)》,批准多个使用境内真实世界数据的医疗器械产品,建设海南博鳌乐城先行区医疗器械临床真实世界数据研究平台,运行真实世界数据应用前置沟通机制等。
4、相关思考及探讨
学习贯彻习近平新时代中国特色社会主义思想主题教育开展以来,器审中心提升科学审评能力调研组问策于实践,取经于实践,就如何编制准确性高、规范性好、实用性强、满意度优的指导原则,进行深入调研、积极思考。下一步,建议技术审评机构紧紧围绕国家药监局“提前介入、一企一策、全程指导、研审联动”的要求,牢牢把握好鼓励创新发展和保障科学审评的关系,通过有效运行指导原则体系,加强指导原则全链条管理,加快指导原则制修订频率,优化指导原则结构布局,对接国际指导原则进行转化。
(一)深化落实“放管服”要求,加强指导原则全链条管理
全链条管理是一种思维方式。通过有效运行指导原则体系,把握并串联指导原则立项、编制、审校、发布、培训、使用、评估、修订等全环节,提高指导原则的质量和价值。
在立项环节,以揭榜挂帅形式确定指导原则编制主体;在编制环节,明确各类指导原则的格式和内容要求,重视研发机构、临床机构、检测机构等各方反馈意见;在审核环节,运行分技术委员会集体审校机制,试行第二类指导原则专家评审机制;在发布环节,上报国家药监局审核同意后简化发布流程;在培训环节,加大云课堂录制力度,进行宣贯;在使用环节,增加指导原则关键词标签,以分类目录为架构搭建指导原则查询下载平台;在评估环节,发布五年以上的指导原则均列入评估范围;在修订环节,综合运用评估结果建立快速修订、微小修订、合并修订和全面修订模式。
(二)鼓励医疗器械创新,加快指导原则制修订频率
当前,医疗器械的创新速度加快,人工智能、生物材料、3D 打印等新技术涌入医疗器械行业,“主动健康”“远程诊疗”“组合产品”“脑机接口”等多学科融合的医疗器械持续引发关注。与此同时,真实世界证据、计算机建模、器官芯片、类器官等新方法逐渐取代传统临床疗效的确认方式,降低了医疗器械的研发成本,进一步加快创新产品上市步伐[2]。
在器审中心提升科学审评能力调研组开展的调研座谈过程中,多位医疗器械生产企业的研发人员、有意进行产品转化的临床医生,均表示希望尽快发布适合本领域的、提供解决路径的、指导价值高的指导原则,认为这便是不断创新的“助推剂”和抵御风险的“压舱石”。国家药监局“十四五”以来发布的多项指导原则定位于国家重点研发计划、中国药品监管科学行动计划、重点领域创新任务揭榜挂帅项目的成果产出。下一步更是需秉持“一企一策”的服务理念,及时制定未上市产品的审评要点,加大创新产品指导原则的发布力度,第一时间修订与产业发展脱节的指导原则内容,依托前沿科技攻关,进行审评重心前移,协同加快产品上市步伐。
(三)以学促干提升科学审评能力,优化指导原则结构布局
单从数量看,现阶段我国制定发布的通用类指导原则仅占10%,但正是占比10% 的指导原则,在解读监管法规、阐释审评理念等方面发挥了关键的作用。安全和性能基本原则、受益- 风险判定、真实世界数据、附条件批准等相关指导原则揭示了科学审评的新方法,临床评价、临床试验设计、接受境外临床试验数据、等同性论证等相关指导原则成为科学审评的新工具,网络安全研究、动物试验研究、注册单元划分、技术要求编写、体外诊断试剂说明书编写等相关指导原则组成了科学审评的新标准。
器审中心通过制定指导原则“十四五”规划和年度编制计划,正在逐步提高通用类指导原则的比例,完善产品类临床评价指导原则。目前,医疗器械说明书编写、真实世界研究设计和统计分析、人因设计、可靠性研究、体外诊断试剂主要原材料、稳定性研究、变更注册等指导原则正在加紧编制,拟于近期发布。真实世界研究试点品种、计算机辅助诊断软件、心脏封堵器、HIV 检测试剂、肿瘤个体化治疗相关基因突变检测试剂的临床研究指导原则也已立项,将有效补充对相关产品设计开发和临床确认的指导。
(四)推进全球医疗器械法规的监管信赖,吸收转化国际指导原则
2019 年6 月24 日起,国家药监局正式启用医疗器械注册电子申报信息化(eRPS)系统,开始实施电子申报制度。通过转化国际医疗器械监管机构论坛(International Medical Device Regulators Forum,IMDRF)注册申报工作组(Regulated Product Submission,RPS) 文件[15],编制了《医疗器械注册申请电子提交技术指南(试行)》,成为全球首个实现全项目、全流程注册申报电子化的国家[16]。在之后《医疗器械监督管理条例》配套规范性文件的修订过程中,以注册申报资料电子目录(Table of Contents,ToC)为基础,明确各目录的注册申报资料要求。IMDRF 管委会各成员国逐步认识到其应用价值,积极推动在本国医疗器械注册监管中使用国际统一的ToC,并一致认为其是统一医疗器械产品电子提交格式的基础[17]。
eRPS 系统的上线、ToC 的国际应用均已成为全球医疗器械法规监管信赖的典型案例。审评面向国际,审评出的产品才能走向国际。技术审评机构仍需持续密切关注美国、欧盟、日本等成熟医疗器械监管机构的指导原则发布进展,根据指导原则体系的现有特点进行规划、布局。据不完全统计,IMDRF 现行指导原则44 项,全球医疗器械监管法规协调会(Global Harmonization of MedicalDevice Regulation,GHWP)制定指导原则56项,共计100 项的国际医疗器械监管共识文件可为成员所用,供成员进行参考、转化和采用。
5、结 语
我们应积极应对医疗器械新技术、新产品带来的新挑战,坚持审评科学理念,面对科技创新发展趋势、国际形势风险考验,制定和转化医疗器械上市前安全性、有效性评价的新工具、新方法、新标准,努力提升指导原则的科学性、前瞻性和引领性。指导原则与审评能力相辅相成,指导原则体系让两者形成良性循环。通过制定指导原则、运行指导原则体系,技术审评机构的审评人员方能在特定领域拥有深厚的专业知识和技能,技术审评机构的审评能力方能对行业重点领域进行深入研究和理解,技术审评机构的审评结论方能具有国际认可度和影响力。
引用本文
高国彪,仉琪,张世庆,姜雨萌.构建医疗器械指导原则体系 筑牢注册技术审评科学基础[J].中国食品药品监管,2023(10):8-17.