您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-07-15 14:28
医用口罩包括医用防护口罩、医用外科口罩和一次性使用医用口罩。依据《医疗器械分类目录》及医疗器械分类界定结果汇总,管理类别为Ⅱ类,外科口罩分类编码为14注输、护理和防护器械-13手术室感染控制用品-04外科口罩;防护口罩分类编码为14注输、护理和防护器械-14医护人员防护用品-01防护口罩;一次性使用医用口罩分类编码为14注输、护理和防护器械-14医护人员防护用品。
一、医用口罩的结构组成和工作原理
1、结构组成
1.1外科口罩通常由面罩、定形件、束带等组件加工而成,一般由非织造布材料制造而成。通过过滤起到隔离作用。
1.2防护口罩通常由一种或多种对病毒气溶胶、含病毒液体等具有隔离作用的面料加工而成的口罩。在呼吸气流下仍对病毒气溶胶、含病毒液体等具有屏障作用,且摘下时,口罩的外表面不与人体接触。
1.3一次性使用医用口罩通常用无纺布或无纺布复合材料制成,可为二层或三层结构,可有可塑性鼻夹,口罩带可为弹性或非弹性,具有过滤颗粒物和细菌等特性。
2、工作原理
医用口罩大部分为自吸式过滤口罩,其工作原理是使含有害物的空气通过口罩的滤料过滤后再被人吸入或者呼出。(见图1):
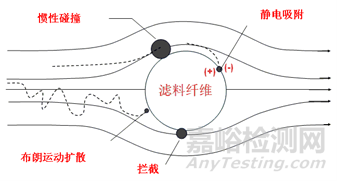
图1 滤料纤维过滤机制示意图
2.1扩散沉积:粒子布朗运动扩散位移到过滤纤维,受分子引力作用而被吸附。最易捕捉小尺度粒子、细纤维和低速运动的粒子。
2.2截留沉积:随气流运动的较大粒子被过滤材料的机械筛滤作用截留。粒子直径与滤膜纤维的直径的比率影响拦截效率。
2.3惯性沉积:粒子通过过滤材料弯曲的网状通道时,粒子由于惯性作用脱离气流撞击过滤纤维,并受分子引力作用被截留。大粒子、高密度、速度快时截留效果好。
2.4静电吸引沉积:粒子被过滤纤维的静电作用产生的沉积。
颗粒越小时,1、4沉积效应越强,颗粒越大时,2、3效果越好,所以并非越小的颗粒越难被过滤。综合4种过滤机制的协同作用,普通机械性滤料最易穿透粒径的范围是0.1µm~0.3µm(见图2)。
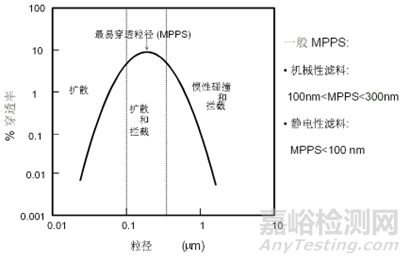
图2 滤料穿透率和粒径关系
二、医用口罩的主要风险
医用口罩主要危险举例见表
表 医用口罩主要危险举例
|
危害类型 |
可能产生的危害 |
形成因素 |
控制措施 |
|---|---|---|---|
|
生物学危害 |
生物污染 |
产品没有消毒/灭菌或消毒/灭菌没有达到标准 |
严格控制消毒/灭菌工艺有明确的消毒/灭菌程序,每批进行消毒/灭菌效果检验 |
|
生物相容性 |
生产引入的外来有害物质没有被有效去除;环氧乙烷残留量超标 |
原材料入厂检验; 严格控制灭菌工艺 |
|
|
与产品使用相关的危害 |
不适当的标签 |
产品最小包装标记不清晰、不全面、不正确 |
标记印刷清晰正确; 标记内容按相关要求标记全面 |
|
说明书上的注意事项不全 |
如缺少详细的使用方法、缺少必要的警告说明 |
规范说明书 |
|
|
对一次性使用产品的很可能再次使用的危害性警告不适当 |
说明书中未包含只限一次性使用 |
规范说明书 |
|
|
功能失效引起的危害 |
不适当的预期用途表述 |
说明书中未能清楚表明产品用途 |
规范说明书 |
|
不适当的产品包装 |
生产、运输、搬运和储存过程中导致包装破损; 包装封口不严密; 包装材料选择不适当 |
严格控制包装工艺 |
|
|
失去产品的完整性 |
产品各构件之间缝制、粘合不严密或材料本身存在破损达不到隔离要求 |
严格控制生产工艺、产品检验 |
|
|
产品对环境的危害 |
环境污染 |
生产环境污染产品,如外来的纤维、粉尘、细菌等其他杂质; 产品原材料受到污染; 储运环节污染产品 |
严格控制生产环境; 严格控制原材料采购、检验; 严格控制产品储运环节 |
三、医用口罩性能研究实验要求
1、化学和物理性能研究
应当明确产品化学、物理和/或机械性能指标的确定依据,所采用的的标准或方法。
2、生物学特性研究
生物相容性评价研究应当明确生物相容性评价的依据和方法;产品所用材料与人体接触的性质;实施或豁免生物学试验的理由;对于现有数据或试验结果的评价。
结合产品特性各部分关注的内容如下:
2.1生物相容性评价的依据和方法:应参照GB/T 16886.1《医疗器械生物学评价第1部分:风险管理过程中的评价与试验》对成品中与患者直接接触的材料的生物相容性进行评价。
2.2产品所用材料及与人体接触的性质:需明确产品所用材料;明确本产品与人体的接触方式、接触时间及接触部位。
2.3实施或豁免生物学试验的理由:产品生物学评价终点建议至少考虑细胞毒性、致敏反应、皮肤刺激或皮内反应。
2.4对于现有数据或试验结果的评价。
3、灭菌工艺研究
医用口罩可以无菌及非无菌形式提供。无菌提供时参考GB 18280.1《医疗保健产品灭菌辐射第1部分:医疗器械灭菌过程的开发、确认和常规控制要求》、GB 18279.1《医疗保健产品灭菌环氧乙烷第1部分:医疗器械灭菌过程的开发、确认和常规控制的要求》和GB/T 16886.7《医疗器械生物学评价第7部分:环氧乙烷灭菌残留量》等相应规定,明确产品包装及灭菌方法选择的依据,经过确认并进行常规控制,并应开展以下方面的确认:
3.1产品与灭菌过程的适应性:应考察灭菌/灭菌方法等工艺过程对于医用口罩产品的影响。
3.2包装与灭菌过程的适应性。
3.3应明确灭菌工艺(方法和参数)和无菌保证水平(SAL),并开展灭菌确认。无菌保证水平(SAL)应达到1×10-6。
3.4残留毒性:若灭菌使用的方法容易出现残留,如环氧乙烷灭菌,应当明确残留物信息及采取的处理方法,并开展环氧乙烷解析的研究。
4、稳定性和包装研究
货架有效期验证试验通常可分为加速稳定性试验和实时稳定性试验两类。加速稳定性试验方案设计可参考YY/T 0681.1《无菌医疗器械包装试验方法第1部分:加速老化试验指南》标准进行,其中“室温或环境温度(TRT)”应选择能代表实际产品储存和使用条件的温度,建议采用保守值设计。在进行实时老化试验设计时,应根据产品的实际运输和储存条件确定适当的温度、湿度、光照等条件。
加速稳定性试验确定的有效期必须通过产品正常储存和使用条件下的实时试验结果进一步验证。如果注册时开展的是加速稳定性验证,开发人需继续进行实时稳定性试验。当加速稳定性试验结果与其不一致时,以实时稳定性试验结果为准。
货架有效期验证试验需采用与常规生产相同的产品进行。验证项目包括产品自身性能验证和包装系统性能验证两方面。前者需选择与医疗器械产品有效期密切相关的物理、化学检测项目,涉及产品生物相容性可能发生改变的医疗器械,需进行生物学评价。包装系统性能验证包括包装完整性、包装强度和微生物屏障性能等验证项目。其中,包装完整性验证项目可包括在设定的时间间隔点目力检测产品包装(是否污染、破损等)及标签(完整性、粘附牢固度、印刷内容清晰度等)、泄漏试验等;包装强度测试项目包括软性屏障材料密封强度试验、无约束包装抗内压破坏试验和模拟运输试验等。
运输稳定性。应当开展运输稳定性和包装研究,证明在生产企业规定的运输条件下,运输过程中的环境条件(例如:震动、振动、温度和湿度的波动)不会对医疗器械的特性和性能,包括完整性和清洁度,造成不利影响。
5、其他
医用口罩产品列入《免于临床评价医疗器械目录》,开发人应当按照《列入免于进行临床评价医疗器械目录产品对比说明技术指导原则》,从基本原理、结构组成、性能、安全性、适用范围等方面,证明产品的安全有效性。

来源:嘉峪检测网


