您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-10-31 17:45
前言
寡核苷酸是一类单链或双链的小合成核酸聚合物,可作用于基因表达水平进而调节蛋白质功能,以达到治疗疾病的目的。继化学小分子和单克隆抗体药物之后,寡核苷酸药物因其在RNA水平上调控疾病基因转录翻译的独特机制,可以填补一些罕见、难治愈并还未攻克的疾病治疗领域,其研发得到了快速发展。近年来,全球已有14个寡核苷酸药物获得上市批准,70多个寡核苷酸药物II期或III期临床试验正在进行中,覆盖罕见病、难治愈疾病等多个治疗领域[1]。
与传统小分子药物一样,寡核苷酸药物在临床前研究阶段,通常需要进行体外代谢稳定性研究。目前所有已上市的寡核苷酸药物均在申报材料中提交了药物的体外代谢研究报告,以阐明药物的代谢能力。本文主要介绍了寡核苷酸药物的代谢酶、体外代谢研究方法以及研究策略,为寡核苷酸新药研发提供参考。
1、寡核苷酸药物简介
寡核苷酸药物(Oligonucleotides, Oligo)是由人工化学合成的12~30个核糖寡核苷酸单链或双链组成的一类药物,通过Watson-Crick碱基配对原则作用于目标信使RNA (mRNA)[2],主要分为反义寡核苷酸药物(Antisense Oligonucleotides, ASOs)和小干扰RNA药物(Small Interference RNA, siRNA)两大类。作为新一类药物分子,寡核苷酸药物极性大、带电荷、需要借助化学修饰和递药系统改善成药性,因而具有不同于化学小分子的药理学特征。未经化学修饰的寡核苷酸药物成药性通常不理想,它们具有较差的PK特性,比如稳定性差,易被核酸酶降解;极性大,对目标RNA的结合亲和力不佳。为了达到临床效用,寡核苷酸必须经过化学改造。
细胞内递送:
与分子质量约7kDa的ASOs相比,分子质量约14kDa的双链siRNA因为较大分子体积和亲水性,经细胞摄取有限。为了增加细胞摄取,siRNA通常连接靶向配体,比如N-乙酰半乳糖胺(N-acetylgalactosamine, GalNAc)或包裹在脂质纳米粒中。(具体递送策略可参考公众号:寡核苷酸药物的“智能”递送。)
作用机制:
寡核苷酸药物通过Waston-Crick 配对,以高选择性、亲和性和目标RNA结合,利用内源性核酸酶降解目标RNA,或通过立体阻断核糖体机制调节RNA剪接和翻译过程。目前,临床在研的siRNA有10多个采取纳米颗粒包裹运载,更多的是采取siRNA-GalNAc技术。(具体作用机制可参考公众号:寡核苷酸药物的代谢及代谢产物分析和鉴定研究。)
2、寡核苷酸药物的体外代谢研究方法
已上市的寡核苷酸药物主要被血浆和组织中广泛存在的核酸内切酶和核酸外切酶所代谢,而不是通过肝脏的Ⅰ相和Ⅱ相代谢酶代谢[3]。核酸外切酶作用于链的末端,导致单核苷酸的释放,而核酸内切酶则在链内裂解,产生不同长度的链。例如,ASOs随血流循环至全身,超过80%的药物分布在肝脏和肾脏,主要经血液和组织中的核酸外切酶和内切酶代谢[4],原型和核酸酶代谢产物主要经尿液排泄[5]。siRNA主要分布器官同样为肝脏和肾脏,与大多数ASOs药物不同,siRNA药物除了需要化学修饰避免核酸酶降解,还需要递药系统的庇护进入靶器官,免于被肾脏排泄和核酸酶降解。
由于寡核苷酸药物特殊的作用机制,以及在细胞水平上发挥基因沉默作用的过程的复杂性,对该类药物提出了新的挑战。
2.1肝匀浆代谢稳定性实验
目前上市的寡核苷酸药物大部分被肝脏迅速摄取,小部分分布在其他组织,在肝脏或其他组织中经由核酸酶代谢。肝组织匀浆液(肝匀浆)是通过将肝组织破碎并均质化得到的,方便获取、储存和运输,它含有肝组织中的各种药物代谢酶。肝匀浆可用于寡核苷酸药物的代谢研究,通过将药物加入肝匀浆中,可以在实验室条件下模拟药物在体内的代谢过程。实验中孵育液的pH、终止方法和匀浆浓度都对匀浆代谢结果具有一定的影响,故将从以上三个方面对肝匀浆代谢模型进行探索。
孵育液pH对匀浆代谢结果的影响
Patisiran分别在孵育液pH为5.0、6.0和7.0的条件下,与人肝匀浆在水浴中孵育适当的时间(如1、2、4、6和24小时)后,左图反义链(AS)和右图正义链(SS)代谢稳定性结果如图1所示。结果表明,随着pH的升高,化合物Patisiran反义链和正义链的代谢速率降低。
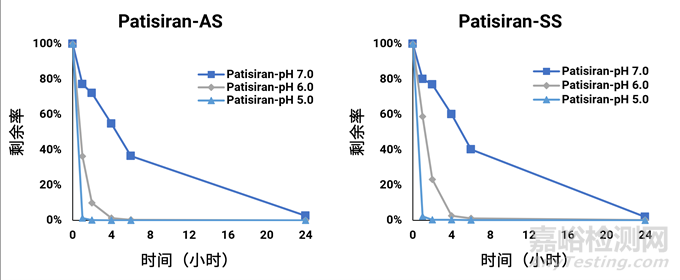
图1. Patisiran在不同pH的人肝匀浆中的代谢稳定性
Inclisiran和Vutrisira在不同pH条件下呈现同样的趋势,如Vutrisira在孵育液pH为5.0、6.0和7.0的条件下,与人肝匀浆在水浴中孵育24小时后,正义链的剩余率分别为16.1%、23.0%、106.7%。
终止方式对匀浆代谢结果的影响
Patisiran与人肝匀浆水浴孵育24小时,分别用OTX (SPE)、NP40 (SPE)和NH4OH (LLE)三种方式进行反应终止以及样品处理[6, 7],实验结果如图2所示。结果表明,三种终止方式的终止效果无明显区别。相较于固相萃取(SPE),液相萃取(LLE)耗材成本较低,优先使用该方法。
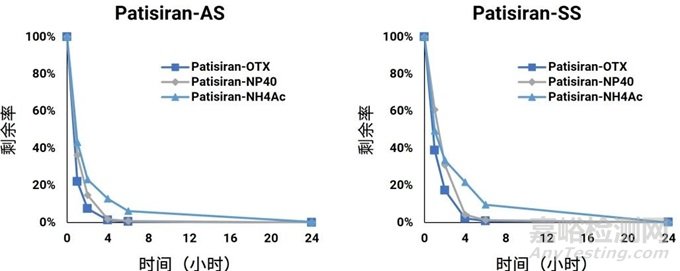
图2. Patisiran在不同终止方式的人肝匀浆中的代谢稳定性
匀浆浓度对匀浆代谢的影响
0.1 mg/mL、0.5 mg/mL、1.0 mg/mL、2.0 mg/mL和 5.0 mg/mL 的五种人肝匀浆与 Patisiran水浴孵育24小时,实验结果如图3所示,结果表明,Patisiran在不同浓度的人肝匀浆中,其代谢趋势略有区别。
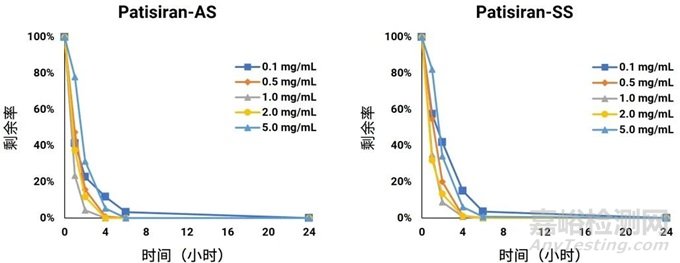
图3. Patisiran在不同浓度人肝匀浆中的代谢稳定性
测试化合物在人肝匀浆中的代谢趋势
测试化合物Patisiran、Inclisiran、Vutrisiran在人肝匀浆酸性条件下,37℃水浴孵育24小时,实验结果如图4所示,测试化合物在人肝匀浆中有明显代谢趋势。Patisiran反义链和正义链均有较明显的代谢趋势,Inclisiran和Vutrisiran的正义链代谢速度明显快于反义链。
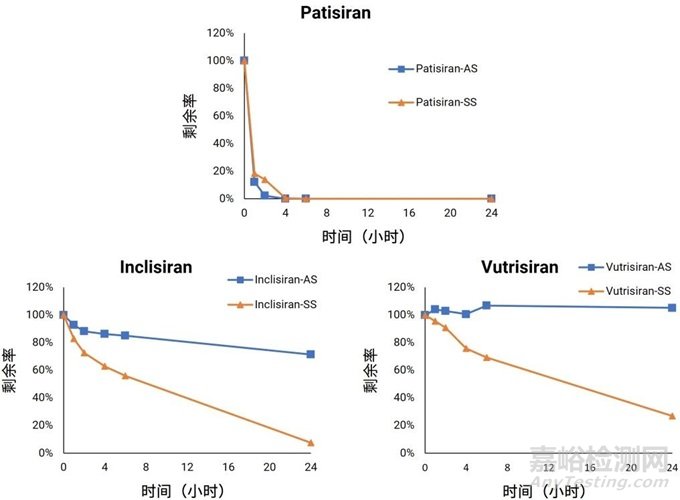
图4. 测试化合物在人肝匀浆中的代谢稳定性
综上所述,我们建立了肝匀浆的模型用于评估寡核苷酸药物的体外代谢。
2.2血浆代谢稳定性实验
血浆作为一种常用的体外基质,方便获取、储存和运输,可模拟药物在体内循环系统的稳定性,因此常被广泛用于药物开发的各个阶段。血浆中含有丰富的核酸酶,所以在寡核苷酸的研究中评估血浆稳定性至关重要。通过对实验设计进行不断优化,采用两个商品化siRNA与大鼠、猴和人血浆;5个商品化ASOs与小鼠、猴和人血浆分别孵育24小时之后,将其剩余量和文献数据进行对比,发现具有较高的一致性。实验结果如表格1和2中所示。
表1. 5个商品化ASOs孵育24小时的剩余率数据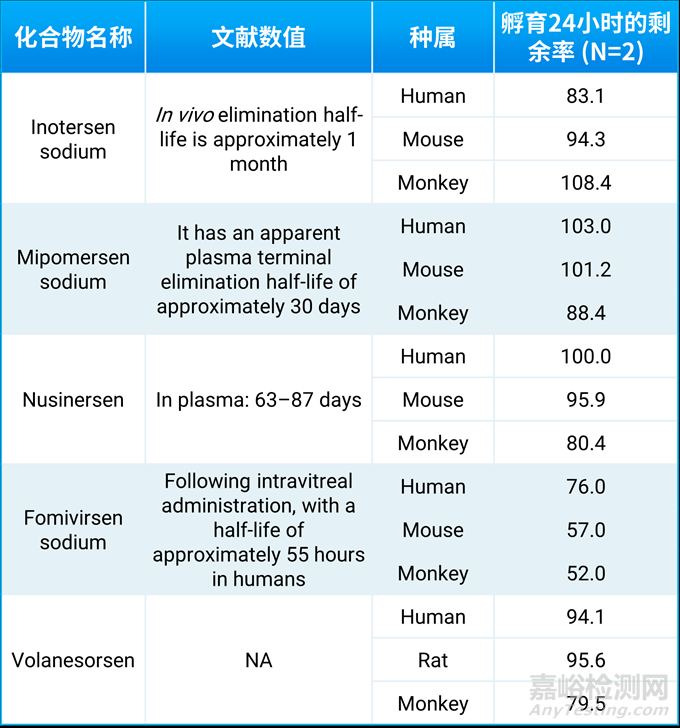
表2. 2个商品化siRNA孵育24小时的剩余率数据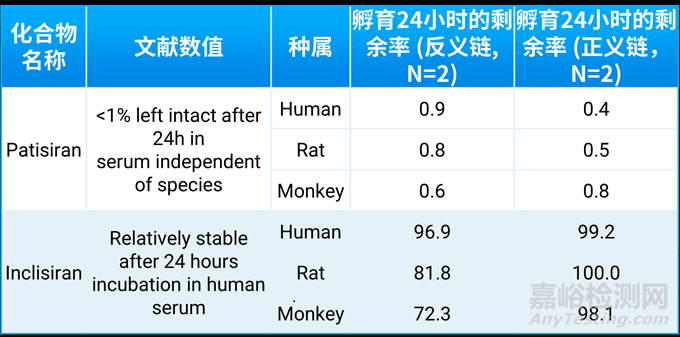
通过以上体外剩余率数据和体内的剩余率或者半衰期数据对比,可以看出体内体外的一致性,寡核苷酸可以通过体外血浆稳定性实验来评估体内循环系统的稳定性。
2.3肝S9代谢稳定性试验
按照肝匀浆代谢模型探索条件进行肝S9试验设计,以确定最终孵育条件。
测试化合物Patisiran、Inclisiran、Vutrisiran在人肝S9酸性条件下,37℃水浴孵育24小时,实验结果如图5所示,测试化合物在人肝S9中的代谢趋势和在人肝匀浆中的代谢趋势具有较高的一致性。
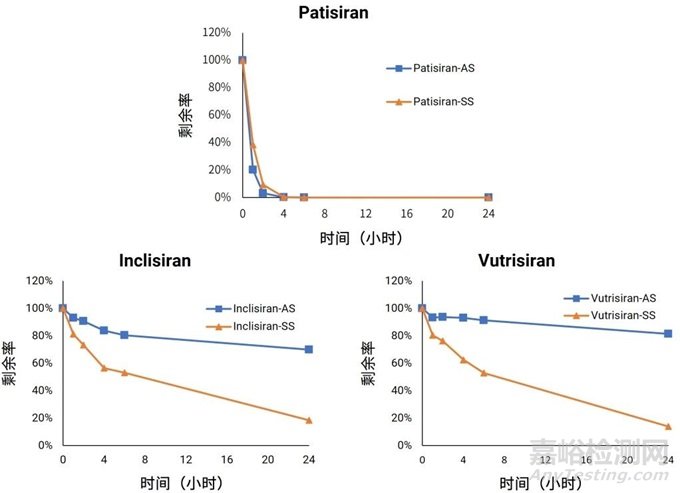
图5. 寡核苷酸在人肝S9中的代谢稳定性
2.4其他基质的代谢稳定性试验
寡核苷酸药物一般基于平台进行开发,体外代谢研究同样基于寡核苷酸药物的结构特点,选择不同的体系。按照肝匀浆代谢模型探索条件进行其他基质的试验设计,以确定最终孵育条件。
3、寡核苷酸药物的代谢研究策略
和传统小分子药物一样,在药物临床前研究阶段,通常需要进行体外的代谢稳定性研究,以评估寡核苷酸药物的稳定性。充分理解该类药物的代谢特性,有助于早期临床试验设计更合理,加快该类药物的上市。一般体内给药后,寡核苷酸药物先进入血液循环,然后在肝、肾等器官中发生代谢和排泄。目前上市的寡核苷酸药物大部分被肝脏迅速摄取,小部分分布在其他组织,在肝脏或其他组织中经由核酸酶代谢,肝脏作为主要代谢器官,在寡核苷酸药物体外稳定性研究中占主要部分,其次是肾脏。
经过实验研究发现,通过体外血浆中孵育,可获得寡核苷酸药物在血中水解酶或核酸酶作用下的代谢稳定性情况。通过肝匀浆孵育,可获得与体内靶组织相近的代谢稳定性行为。同时,通过体外模型如S9、溶酶体等可进一步评估寡核苷酸药物在靶组织中的稳定性。
参考文献:
[1] Oligonucleotide Therapeutics: From Discovery and Development to Patentability;Pharmaceutics 2022, 14, 260
[2] Bennett CF. Therapeutic antisense oligonucleotides are comingof age [J]. Annu Rev Med, 2019, 70: 307-321
[3] Andersson, P. & Den Besten, C. CHAPTER 20: Preclinical and Clinical Drug-metabolism, Pharmacokinetics and Safety of Therapeutic Oligonucleotides. RSC Drug Discov. Ser. 2019-January, 474–531 (2019)
[4] Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism [J]. Expert Opin Drug Metab Toxicol, 2009, 5: 381-391.
[5] Mustonen EK, Palomaki T, Pasanen M. Oligonucleotide-based pharmaceuticals: non-clinical and clinical safety signals and nonclinical testing strategies [J]. Regul Toxicol Pharmacol, 2017, 90: 328-341.
[6] Molecular Therapy: Nucleic Acids Vol. 21 September 2020. 725~736
[7] Metabolism of Antisense Oligonucleotides in Rat Liver Homogenates; JPET 292:140–149, 2000

来源:药明康德DMPK


