您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-02-10 14:52
人体吸收、分布、代谢、排泄(hADME)研究主要目的有两个:1)鉴定和定量外周血中的原型药及代谢产物;2)定量表征药物消除特征。
hADME起源
hADME与George de Hevesy最初提出并建立的同位素示踪剂的使用有关,其本人并因此获得了1943年诺贝尔化学奖。Hevesy早期工作将放射性铅(210Pb和212Pb)用于化学和生物学研究,实现了放射性元素作为示踪剂的应用。将放射性同位素,通常是氚(3H)或碳-14(14C)或稳定同位素(2H、13C、15N、18O等)掺入参与化学反应或生物转化的底物或中间体中,可以追踪标记的中间体或产物,既可定性,又可用于定量。
Hevesy对示踪剂的使用很快被其他人采用,并最终扩展到代谢和生物化学的研究。比如使用14CO2和3H2O探索光合作用,使用稳定和放射性同位素标记的化合物确认Krebs cycle,以及使用32P和35S确定遗传物质是DNA,而不是蛋白质。
虽然3H于1934年就被发现,但其在核武器开发中的使用限制了其在20世纪40年代和50年代的可用性和研究用途。碳元素方面,早期的生化研究采用的是短半衰期(20.4分钟)的11C。不过,1940年发现了长半衰期的14C(5730年),并在生化研究中得到了更广泛的应用。采用Berkeley辐射实验室获得的14C进行了[14C]二苯并蒽的放射性化合物合成,并于1948年首次发表了14C标记二苯并蒽在动物体内吸收、分布、代谢和排泄研究中的应用。这项研究在收集和表征排泄物和胆汁中14C的消除方面与现代ADME研究相似。
14C标记化合物的人体ADME研究最早是Alpen等人于1951年披露,研究的14C标记水杨酸的代谢。不过,在那个年代,原型药和代谢物的分离方法、鉴定方法和检测方法比较受限。生物样本主要收集的是外周血和尿液,很少关注粪便中的原型药物及代谢产物。
很难确定放射性标记ADME成为常规研究的具体时间,普遍认为这个时间点在20世纪50年代初至中期。到了20世纪70年代初,hADME研究已经非常普及。
20世纪50年代末气相色谱质谱法的发展,20世纪70年代和80年代出现了高效液相色谱法(HPLC),21世纪初出现了超高效液相色谱仪,使得药物和代谢物的分辨率更高,分析速度更快。此外,液相色谱(LC)分离方法与热喷雾界面和电喷雾界面结合,以及核磁共振仪器的改进,对代谢物的结构鉴定产生了巨大影响。hADME研究中的样品分析和代谢物结构测定受益于分析方法和仪器的进步。
标准ADME设计
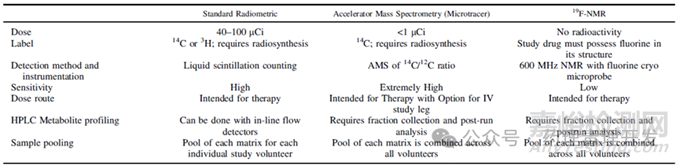
人体ADME的设计在过去数十年基本没有太大变化。临床研究志愿者给予14C标记化合物,其中的14C通常放置在代谢稳定的位置,以避免放射性的丢失。14C的剂量需要足够高,以保证液闪仪(LSC)可以对原型药和代谢物的准确定量,通常在40至100 μCi之间。给药途径与临床拟用途径一致,通常是口服。14C标记药物ADME通常是单次给药,使用的制剂通常不是最终或商业化制剂,而是专门为本研究制备的溶液或混悬液。因此,获得的药代动力学特征可能并不能精确模拟真实片剂或胶囊制剂。
在给临床试验受试者(通常为4-8人)服用后,在设定的时间间隔内收集尿液和粪便样本。收集的持续时间根据估计的药物及其相关物质的排泄特点进行预设,也可以通过实时分析样本和数据进行灵活设置。当达到预定的回收率(通常为90%)或药物相关物质的排泄率降至预定阈值以下(如一天内1%)时,受试者可以退出研究。LSC可以在短时间内完成检测,以实现数据驱动的及时决策,如样本采集周期。血液样本也被收集,用于测定总药物相关物质的药代动力学,并与原型药的药代动力学进行比较。
尿液的分析比较常规、简单,从尿液中取出小份样品,直接LSC检测,再根据取出的体积占比,折算某给药间隔内总尿液放射性。所有给药间隔尿液总放射性相加,再除以给药总放射性,就是尿液排泄的药物比例。粪便样本操作起来有点复杂,因存在颗粒或者有色物质,可能会干扰检测,通常会将粪便样本高温烧成CO2,再行测量。其它生物样本的收集和检测,如呼出的空气、汗液,并不常见,但不排除有些药物通过这些途径排出体外,需根据具体情况决定是否收集和分析。
血浆也会进行总放射性分析。当然,有时用全血。血浆可以直接测量,全血则需要做些预处理如匀浆或漂白等。在经典的人体ADME研究中,原型药物也会采用非放射性方法定量测定(常用的是HPLC-MS),获得Cmax、Tmax、AUC、t1/2等参数,可以与放射性方法测定的相应参数进行比较。
除了吸收和排泄,还会对血浆、尿液和粪便匀浆中的代谢物谱进行定量测定。为减少代谢物的损失,通常前处理比较简单,采用简单的液相提取,通过离心去除盐和蛋白质,蒸发含有药物相关物质的上清液,再重新复溶,然后进行HPLC分析。整个样品处理过程,总放射性的回收率应为90%或更高,以增加特定代谢物未丢失的可信度。代谢物被色谱分离成离散的峰,这些峰可以通过LSC进行定量(可以通过离线或在线两种形式),同时将一部分HPLC洗脱液转移到质谱仪,以获得代谢物的结构信息。最终实现代谢物的结果鉴定和定量分析。
当然,关于代谢物检测,并不是对每个样本均进行分析,无论血浆、尿液还是粪便,都会进行样本合并,对合并后的单一样本进行分析。需要注意的是,必须按比例考量每个样本的体积/重量,以生成真正代表总排泄量的分析样本。
新技术应用—加速器质谱技术(AMS)和核磁共振(NMR)
AMS技术是一种高灵敏度的核分析技术,出现在20世纪70年代,但直至过去10年,其大小和成本才开始满足小型实验室的使用。相较于传统的LSC,AMS的检测灵敏度提高了多个数量级,直接的优势是可以降低给药量,100-1000nCi即可。事实上,基于AMS的ADME研究中,血浆和排泄物样本中的14C含量已经很低,不再视为具有放射性。极低的电离辐射暴露对受试者没有安全性风险,正因如此,用于进行人体组织剂量估计的动物定量全身放射自显影研究(QWBA)不再是进行人体ADME研究的先决条件。当然,有利也有弊,AMS相较于LSC,样品制备过程更久、更费力、设备成本也更高。
14C标记化合物需要专门制备样品,时间可能长达几个月,成本不菲,高则数十万美元。氟存在于许多药物中,与质子相比,没有内源性含氟干扰。因此,氟核磁共振(F-NMR)可用于含氟药物的ADME,不需要14C标记。当使用F-NMR进行ADME研究时,样品处理程序虽然不如LSC简单,但比AMS有优势。与LSC和AMS相比,NMR的挑战在于灵敏度较低,即使使用高频仪器(>500 MHz),也需要对大量样品进行处理和浓缩才能可靠定量。Pearson等人于2019年首次报道了使用F-NMR对leniolisib进行hADME研究。
下表比较了传统标准放射性检测、AMS和NMR不同技术路线用于hADME研究的特点。
动物ADME研究
目前常见做法是,在进行hADME研究之前,开展至少一项放射性标记的动物ADME研究。动物ADME研究的路线通常是先开展大鼠的ADME研究,然后再进行第二个毒理学种属的ADME试验。随后进行QWBA,该试验可用于组织剂量计算,从而为后续hADME研究的放射性剂量提供依据。另外,动物ADME研究还可以为hADME研究提供其它支持,如基质的提取技术、分离代谢物的色谱系统和代谢物结构表征。而且,动物研究可以更具侵入性,例如胆汁插管大鼠试验。当然,动物ADME特征可能并不能完全反映人体情况。
2012年,Obach等人引发了一场争论,主题是关于动物放射性标记的物质平衡和排泄研究是否仍然必要。争论的核心是,相较于对动物的详尽研究,对人类代谢物的早期了解,可能更为重要。而利用现代技术进行的早期hADME研究(不迟于ⅡA期)是可以鉴定主要的人体代谢物的。可以使用非放射性标记的方法在临床样本和毒理学样本之间进行初步比较,以评估人体代谢物在动物中是否足以覆盖。当然,有些情况需要在动物中进行放射性标记的物质平衡研究(比如评估一种可能对该动物种属造成毒性的种属特异性代谢物,与人体无关)。White等人(2013)认为,在至少一个种属中进行放射性标记的物质平衡研究对药物开发至关重要,因为这是预期监管机构要求的申报资料的一部分,且这些研究可以为后续处理人体样本提供参考。
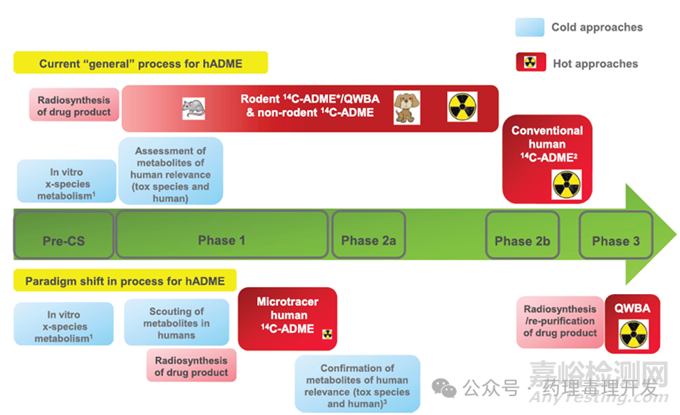
目前,争议依然存在。由欧洲制药工业协会联合会(EFPIA)药物代谢和药代动力学(DMPK)网络赞助的药企联盟,组成了一个工作组,发表了一篇白皮书(Young et al., 2023),其中写到,工业界广为接受的观点是放射性研究不应作为默认的“check boxes”研究内容,即不是所有药物的默认勾选项,而要基于case-by-case分析。最近,abrocitinib被批准用于临床,本品并未在动物中开展任何放射性标记的排泄研究,只进行了hADME和大鼠QWBA研究。与下图箭头下方的路线一致。下图是该白皮书给出的current general process,paradigm shift in process,图中标识了hADME不同试验开展的时间点。箭头上面是目前用的比较多的hADME路线,即主流路线。箭头下面则是白皮书提出的新的human first/human only变化。蓝色为冷药试验,红色为放射性药物研究。二者最大的区别是,箭头下方不需要啮齿类和非啮齿类14C-ADME研究,更依赖于对冷药代谢产物的鉴定,解决代谢产物安全性相关的问题。
人体ADME研究重要性
在hADME研究中,排泄物的主要作用之一是研究物质平衡。物质平衡对有效性和安全性的支持有限,其主要目的是为药物清除提供支持和信心。例如,低回收率可能表明有生物样本遗漏或不完整,药物依然在体内,或者药物或代谢物从呼出的空气中消除。FDA在《Clinical Pharmacology Considerations for Human Radiolabeled Mass Balance Studies Guidance for Industry》指出,回收率至少为90%。如果不符合这一标准,需提供“充分理由”。
大多数药物是通过以下一种或多种机制消除:1)小肠中的代谢/转运;2)肝脏代谢/转运;3)肾小球滤过和肾小管分泌。如果药物消除路径出现损伤,会改变药物的药代动力学行为,甚至需要调整药物的给药剂量。肝、肾损伤患者调整剂量需要考量的因素之一就是每个消除路径的占比,而这一信息就来自hADME研究。
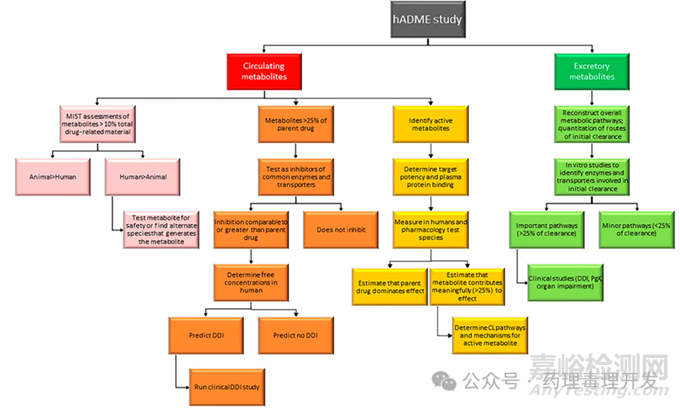
药物在人体中的代谢谱及代谢产物浓度的测定具有重要意义,可以为体外、动物和其他临床研究提供更多信息。代谢物谱的测定对于化合物是否需要开展代谢产物安全性评价也是需要的。对于人体代谢比例超过10%的代谢产物,且动物体内没有或暴露量不能覆盖的情况,根据FDA代谢产物安全性评价指导原则,需要补充开展非临床安全性研究。另外,有些代谢产物是具有活性的,对药效也有贡献。除了药理学活性,代谢产物可能对药物代谢酶或转运体产生与原型药不一样的作用。所以,hADME中对代谢产物浓度和结构的研究可以提供很多数据,为体外或临床药物相互作用研究提供支持。下图比较直观的展示了hADME研究如何支持进一步体外、动物和其它临床研究的。
人体ADME研究的开展节点
有些人依然认为hADME研究在药物开发过程中可以事后补充开展,即等其它研究完成后再行开展,开展节点尽可能的往后推迟。其中一部分原因是Ⅱ期临床具备比较高的失败率,再加上hADME研究费用不菲,想等Ⅱ期临床完成后再行开展。但考虑到DDI风险、代谢产物安全性等问题,hADME研究节点应尽量提前。不同监管机构也给出了各自的答案。
2024年1月,NMPA颁布的《放射性标记人体物质平衡研究技术指导原则》指出,应合理安排放射性标记人体物质平衡研究的开展时机,鼓励在药物临床开发的早期进行放射性标记人体物质平衡研究,以尽早获得必要的信息以支持药物研发总体计划,通常建议在确证性临床研究开始前完成。
2024年5月颁布的ICH M12《药物相互作用研究》指导原则规定,通常应在III期研究开始前获得物质平衡研究的结果。根据物质平衡研究和体外研究的结果,应考虑使用指针酶强抑制剂和诱导剂进行临床研究,以确认主要代谢途径,并确定具有临床意义的DDI风险。
2024年7月,FDA在《Clinical Pharmacology Considerations for Human Radiolabeled Mass Balance Studies Guidance for Industry》指出,物质平衡研究应该早点开展,最迟应在晚期临床开始前完成。FDA认为:1)物质平衡研究可以提供代谢和排泄途径相关信息。这些信息与其他体外和体内数据一起可以为DDI研究提供支持;2)鉴定应进行非临床安全性评估的代谢物;3)指导肾和/或肝损伤研究;4)避免入组时不同肾功能和/或肝功能患者的排除,或为安全性和有效性临床试验中这类患者的用药剂量提供信息。
上文提及的白皮书也提到,各公司对hADME各项研究的开展时间节点看法不一,仅考虑在合适的时间开展。当然,通常也不会在首次人体临床研究中开展,毕竟制备符合GMP级别放射性标记物料成本是非常高的。传统做法是在临床Ⅰ期开展冷药人体代谢产物鉴定、啮齿和非啮齿类14C-ADME及大鼠QWBA研究,Ⅱ期临床阶段开展人体14C-ADME研究,这也是当前的主流做法。随着新技术和新观念的出现,已经有企业开始尝试新的思路,前文已有提及,不再赘述。

来源:药理毒理开发


