您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2018-06-27 09:27
摘要:杂质研究是药物质量研究的重要方面,如何制订杂质限度是比较复杂的问题,本文从审评实践中介绍了药物有机 杂质限度的制定方法,并对不同类别的药物和药物研发不同阶段的有机杂质研究进行了评述。
1 杂质的分类和存在的问题
任何影响药物纯度的物质统称为杂质。在药物研发中杂质的研究贯穿其整个阶段,是保证药品质 量的重要内容。药品中的杂质分类通常有四种方法:
①按理化性质分类 :有机杂质、无机杂质及残 留溶剂。
②按来源分类:工艺杂质 ( 包括合成中未反应 完全的反应物及试剂、中间体、副产物等 )、降解 产物、从反应物及试剂中混入的杂质等。
③按毒性分类 :毒性杂质和普通杂质等。
④按化学结构分类 :其它甾体、其它生物碱、 几何异构体、光学异构体和聚合物等。 杂质的存在对药物的安全性和有效性有很大 的影响,如何制定杂质的限度也是药物研发过程中 一个非常敏感的问题。目前国内在药物杂质研究中 存在的主要问题是“杂质限度制订的依据不充分”。
研制单位通常是根据多批样品的研究结果确定,或凭所谓的“经验”制订。特别是方法学研究不全面, 没有对杂质进行有效地分析,并进行定性研究,导 致杂质研究不完善,成品与国外产品的质量差别较大。
本文重点对药品中有机杂质限度的制订方法进行了讨论。
2 一般要求
药品的开发需遵循三个原则 :安全、有效和质量可控。因此,在制订质量标准中杂质的限度时, 首先应从安全性和有效性方面考虑,尤其对具有药理活性或毒性的杂质,要严格控制其限度。其次, 应考虑生产的可行性及批与批之间的正常波动,以及药物本身的稳定性。在保证安全和有效的前提下, 根据工艺的波动情况制定合理的限度。此外,在药 物研发过程中不同药物或者同一药物在不同研究阶 段的侧重点是不一样的。因此,其杂质研究也各具特点,需要根据这些具体情况分别对待。
通常,可以根据药物规定的剂量确定降解产物的报告阈值、鉴定阈值和界定阈值,以此来确定杂质的限度。另外,亦可通过已知杂质的毒性数据, 或其毒性研究结果来确定杂质的限度。
2.1 ICH的一般要求
在确定了药物的剂量后,可以根据降解产物的阈值来制订杂质的限度。此时,可参照 ICH( 人用 药品注册技术要求国际协调会 ) 的相关要求制订杂质限度 。 如,按 ICH要求,对于新的原料药,每日最大剂量大于 2 g时,单个杂质的界定阈值为 0.05% ; 每日最大剂量不大于 2 g时杂质的界定阈值 0.1% 或 1 mg( 按严格的要求定 )。当一个原料药的每日 最大剂量为 1.5 g 时,如按界定阈值 0.1%计,则 杂质的绝对摄入量为 1.5 g×0.1% =1.5 mg,大于界定阈值 1 mg ;如按阈值 1 mg 计,则杂质的限度值 为 1 mg÷1.5 g×100% =0.07%,与界定阈值 (0.1% ) 不一致。在这种情况下,制订杂质限度的原则是“按严格的要求定”,即杂质限度应订为 0.07%。
当然ICH只是一般的要求,对于特殊杂质 ( 具 有一定活性 ) 或杂质限度超出ICH的要求时,应进行相关的安全性研究或根据文献数据,以判断限度的合理性。
2.2 已知杂质化合物的毒性数据
对于已知杂质化合物,可参考相关的文献,或 一些数据库 ( 如 www.discoveryGate.com 网站 ) 查得其毒性数据,并根据药品临床拟用的剂量推算药 物的安全限度。
比如,杂质苯肼的临床主要毒性反应为急性血管内出血,内脏器官损害包括肝、肾、心、骨髓和中枢神经系统等的多发性损害,高浓度还可引起肺组织损害 ;即使作为苯肼衍生物,也具有潜在的致癌性风险。文献显示,雄性鹌鹑静注苯肼 0.6、 6、60 mg/kg,给药后 24 h 和 72 h 均产生明显的溶血反应,并伴有淋巴细胞增多、单核细胞减少和嗜酸性细胞减少。
因此,对于某些药物中的苯肼杂质, 应进行必要的控制。人体试验或用药中的苯肼总量 应不高于0.05 mg/kg。若人体最大用药剂量为2 g时, 产品中如含有该杂质,按人体重 60 kg 计,其限度值为 0.05 mg/kg×60 kg÷2
g×100% =0.15%。
2.3 根据安全性的研究结果推算
对于没有安全性数据的杂质,需要进行安全性试验,然后根据杂质的NOAEL( 未观察到毒性反应的剂量水平,no adverse effect level) 数据进行推 算。
一般情况可采用以下方法 : ①对单个杂质进行毒理学研究,确定其NOAEL, 制订该杂质的限度。如,某创新药临床前试验样品中杂质A的限 度为 0.34%,每天的最大用量拟定为 10 mg。其临床研究用药,申报单位将杂质 A 限度确定为 0.5%。 这一限度不但超过了 ICH Q3 规定的常规杂质限量 标准 ( 0.1% ),也超过了临床前试验样品中杂质 A 0.34%的限度。
因此,在该品种的审评中,药审中心建议提供该杂质限度确定的依据。研究者对此进行了说明,其推算过程如下 :该药在大鼠重 复给药 6 个月的毒性试验中,无毒性反应剂量为 26.5 mg·kg-1·d-1,则杂质 A 的实际用量为 26.5× 0.34% =0.09 mg·kg-1·d-1。通过动物类比法转换为 0.09 mg·kg-1·d-1×6=0.54 mg·/m-2·d-1( 大鼠在人体中 mg/kg 剂量的转换因子为 6),再用 FDA 转换标准 ( 人体中 mg/kg 与 mg/m2 的转换因子为 37),得出相当于 60 kg 的人体耐受杂质 A 的量为 0.54×60÷ 37=0.88 mg/d。考虑到该新药在临床研究中的最大 给药剂量为 10 mg,若按 0.5%的杂质含量计算, 每日的杂质 A 最大剂量为 0.05 mg/d,仅为杂质 A 无毒性反应剂量 0.88 mg/d 的 1/17,因此杂质 A 的限度订为 0.5%是可行的。
②根据毒理学实验样品杂质含量情况进行推算,制订出杂质的限度如,某个药物口服给药的每日最大剂量为 60 mg,相当于一个体重 60 kg 的人给药剂量为 1mg/ kg。在 13 周犬的口服给药毒理学研究中,确定其 NOAEL 为 2 mg/kg。根据该批样品中杂质 A 的含 量为 0.1%计算,杂质 A 最大安全性限度为 2 mg/ kg×0.1% =2 µg/kg,换算到人体的杂质 A 安全剂量 约为 1µg/kg( 犬在人体中 mg/kg 剂量的转换因子为 1.8),则杂质 A 的限度为 1 µg/kg÷1 mg/kg×100% =0.1%。 以上方法只是一个参考,研究者可根据研究结果综合考虑,选择合理的方法,制定科学、合理的 杂质限度,当然不同类别的药物杂质限度的研究有所不同。
3 对创新药物的要求
如前述,药物在不同研究阶段的侧重点是不一 样的,其杂质研究也各具特点。对于创新药物而言, 其研发是一个长期、复杂的过程,在这个过程中, 随着对化合物的药理、毒理作用和药效等方面研究 的不断深入,研发者会对目标化合物的药用前景不断进行评价和评估,因此在不同的开发阶段对杂质的研究要求也各不相同。
3.1 研发初期
在药物研发初期 ( 一般情况在临床研究Ⅱ a 以前 ),由于对拟开发化合物的前景不明确,原 料药的制备工艺、制剂的处方尚不能最后确定, 且化合物的制备规模也不会太大。因此在进行 CMC(chemical manufacture and control) 的研究时, 主要目的是结合药理毒理的研究结果,保证目标化合物的安全性,同时保证目标化合物的质量基本可控。在此期间,根据临床研究的需要,药物的剂型、 处方工艺、规格等均可能发生变更,因此,该阶段 一般采用“通用”的方法 ( 一般可根据药典的常规 要求 ),质量的控制项目采用常规的控制项目 ( 药 典的常规要求 ),目的是保证化合物质量的基本可控和工艺稳定性 ( 质量一致 ),其杂质限度的制订可根据药理毒理的研究结果,只要保证杂质在安全 范围内即可。
3.2 研发的中、后期
在研发中、后期 ( 一般情况下在临床研究Ⅱ b 以后 ),药物的工艺、剂型和处方已基本确定,样品的产量亦已放大至一定规模,在保证安全性的前提下,可结合大规模生产、成本、稳定性等方面的要求,综合考虑制订一个合理的限度,并对相关的杂质进行定性。原则上,质量标准中杂质的限度, 应不低于 ICH 的一般要求。
某一个创新药杂质在不同研发时期的限度见表 1
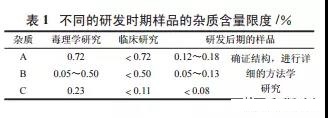
上述数据说明 :①在药物研发的初期,临床样 品的杂质水平不应超过毒理学样品的杂质水平,单 个杂质一般不需要定性。②在研发后期,产品的开发前景已经明确,需要对这些杂质的结构进行确证, 并进行详细的方法学研究,包括对每一个杂质的专属性、灵敏度等的研究。特别是在研发后期,随着生产工艺的优化,规模化样品批产量的质量明显提 高,研究者根据实际情况,最终确定了杂质的限度: 按外标法以峰面积计算,杂质 A、B、C 均不得过 0.2%,有关物质总量不得过 1% ( 一般不得过根据 药品临床用剂量推算的药物安全限度 )。
4 对仿制药物的要求
4.1 仿制国内已上市的药物 ( 已有国家标准 )
这类药物有关物质的限度,主要根据国家标准制订,同时可参考技术先进国家药典的收载情况或 国外产品标准,再结合药物工艺和处方进行必要的研究后制订。
在仿制这类药物时,首先应考虑到国家标准也 需要提高这个问题。特别是上市时间不长的品种, 实际上生产单位在批准生产后有一个逐步完善大生 产工艺、提高和稳定成品质量的过程,即存在着生产单位提出补充申请以修订质量标准的可能。如果被仿制药物已同时被技术先进国家的药典 ( 如美国 药典或欧洲药典 ) 收载了,则应该进行相关的比较。 应注意的是,方法和限度是相对应的,不同的方法对应的限度可能不一致。因此,有关物质检查的方法如果不一样,需要对不同的方法进行互相比较研 究,考察方法的适用性。应特别注意不同方法检出的杂质数量和含量,如有新的杂质产生,其限度的制定可参考前述的方法。
如,抗肿瘤药奥沙利铂 (oxaliplatin),目前国家标准的杂质检查方法相对粗糙,没有对单个杂质进行定性和检查,而且限度的依据不充分,其国家标准需要提高。从奥沙利铂的结构特点分析,可能存在的杂质较多,如草酸、双水合二氨环己烷铂及 其二聚物、二羟合奥沙利铂等,这些杂质需要采用不同条件的 HPLC 方法才能有效地检出。因此研究中在确认了方法的可行性后,可参照技术先进国家药典标准执行,杂质限度亦照此制订。
4.2 仿制国外上市的药物 ( 国内没有上市,没有国 家标准 )
这类药物杂质限度的制订比较复杂,如果能查到国外上市药物的标准情况,杂质限度的制订即可以参照执行。如果没有这方面的数据,需要根据前述的杂质限度制定要求,在进行相关的研究后,再制定合理的限度。 如氯法拉滨 (clofarabine),一种用于治疗儿童 急性粒细胞性白血病的药物,国外已经上市。国内的一些申报单位在仿制该药时,存在有关物质限度不一的问题。有的根据经验确定,有的是根据多批样品的检验结果确定,限度制订的依据不充分。因此在审评过程中,药审中心均给这些研制单位下发了资料补充通知,要求说明有关物质限度的制订依据,并对大于 0.1%的杂质进行深入的研究。
5 结语
杂质的研究是一个复杂的过程,是药物研发中 一个非常重要的环节,是保证药物质量的关键,如何进行杂质研究、限度如何制定以及不同阶段的研 究策略,不同的研发企业有不同的操作方法,但是方法的选择和限度的制定应有充分的依据,这样才能有效的控制产品的质量,以确保用药安全。

来源:AnyTesting


