您当前的位置:检测资讯 > 生产品管
嘉峪检测网 2020-04-24 18:15
随着医疗器械监管法规体系不断完善与规范化,医疗器械生监督检查及飞行检查也将成为日常,医疗器械生产企业应依据医疗器械生产质量管理规范及相关法规要求,定期在企业内部开展生产质量管理体系自查并将缺陷部分进行分析与整改,提高企业生产和质量管理水平,保证生产质量管理体系的有效运行,从容应对监管部门的监督检查或飞行检查。
一、医疗器械体系审查内容
医疗器械体系审查主要关注与产品研制、生产有关的设计开发、采购、生产管理和质量控制等内容,重点核查设计开发、生产等过程数据真实可靠、完整、可追溯。
|
项目 |
主要审查内容 |
|
1. 机构与人员 |
组织机构、相关人员专业背景、资质及工作技能 |
|
2. 厂房、设备与设备 |
产品研发、样品试制相适应的厂房、设施、设备和仪器,满足质量控制要求 |
|
3. 文件管理 |
设计开发及研制原始资料,研究数据的真实性、完整性和可追溯性,研制过程记录如主要物料领用记录、仪器设备使用记录、称量记录、配制记录等,增加研发过程的可追溯性。 |
|
4. 设计开发 |
医疗器械设计开发文档源于设计开发评审、验证、确认和设计转换活动的相关文件,包含设计开发程序、开发计划及建立的记录,保证对历次设计开发最终输出过程及其相关活动的可追溯性。 |
|
5. 采购 |
原辅料合法来源证明,检验报告、采购控制记录及供应商管理。 |
|
6. 生产 |
关键工艺和方法确认,样品生产记录可追溯,确保在实际生产体系下生产样品。 |
|
7. 质量控制 |
样品生产过程检验记录可追溯,样品去向可追溯等。 |
|
真实性核查 |
- 产品注册检验样品真实性 - 临床试验样品真实性 - 样品和生产地址与申报生产地址一致性 - 试生产的样品批次及批号、规格、每批数量、送检样品和临床样品批号及数量、留样品批号及数量、现存样品批号及数量、主要原材料数量等可追溯 - 过程检验原始记录、出厂检验原始记录满足设计输出检验规程与产品技术要求 - 样品台帐、留样记录 - 原材料采购记录 |
二、医疗器械体系审查及整改流程
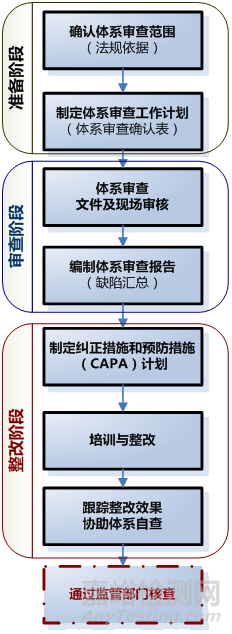
三、医疗器械体系常见缺陷项及整改建议
|
常见缺陷项 |
整改建议 |
|
1.缺少人员培训的相关记录 |
应当进行符合《医疗器械生产质量管理规范》及相关附录中要求的与其岗位要求相适应的培训,留下培训相关记录。 |
|
2.生产管理不到位 |
制定受控的关键或特殊过程的作业指导书,对关键或特殊过程的重要参数进行验证或确认,符合《医疗器械生产质量管理规范》中应当明确关键工序和特殊过程并编制相应规程或指导书的要求。 |
|
3. 质量控制文件缺失或擅自更改、质量控制不到位 |
1)明确描述产品抽验方法及判定规则,符合《医疗器械生产质量管理规范》中要求按强制性标准以及经注册或者备案的产品技术要求制定的产品检验规程。 2)产品检验按产品技术要求的规定执行,符合《医疗器械生产质量管理规范》中对产品检验的相关要求。 3)现场合理摆放产品标识及检验状态标识,符合《医疗器械生产质量管理规范》中对标识的相关要求。 4)制定产品放行程序,明确放行程度、条件和放行标准,符合《医疗器械生产质量管理规范》中对放行的相关要求。 |
|
4.未对质量管理体系的运行进行评价和审核 |
定期对产品质量及质量管理工作进行审核、评审和记录,符合《医疗器械生产质量管理规范》要求定期开展管理评审和内审,以确保其持续的适宜性、充分性和有效性。 |
|
5.部分厂房设施不满足生产条件 |
厂房各区域修建根据区域情况符合《医疗器械生产质量管理规范》及相关附录中要求,满足相应储存条件的要求,满足产品的生产、储存、检验条件,且各区域具备温湿度等环境监视管理设备,产品各部分在对应区域生产、储存、检测。 |
|
6.设计开发管理不到位 |
提供完整的产品设计开发文档,产品输出内容齐全,保持设计开发更改记录并符合《医疗器械生产质量管理规范》中对更改进行识别并保持纪录的要求。 |
|
7.不合格品控制不到位 |
不合格品、待检品和合格品三者均单独设立区域进行标识区分,使其隔离,对应记录完善真实,定期进行评审。 |

来源:CIRS医疗器械监管动态


