您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-06-28 19:29
目前缺少关于医疗器械“服务”和“再制造”之间的区别的澄清性文件,而对于执行再制造的实体是有一定的法规监管责任的。
就此,FDA于2021.6.24发布了医疗器械再制造指南草案,以帮助澄清对器械进行的活动是否为“再制造”,并包括了关于标签中应包含的信息的建议,以帮助确保拟在其使用寿命内使用的设备的持续质量、安全性和有效性,并提供了一些评估示例,以及是否是再制造的评估文档的示例。
1、FDA相关背景介绍
FDA一直在努力获得更多关于“服务”和“再制造”之间区别的观点,重要活动包括以下:
2016年:FDA公开了一份公众意见摘要,并举办了一次公众研讨会。
2018年: FDA发布了" FDA Report on Device Servicing", 讨论并分析了收到的关于“服务”不足导致或促成的评论,投诉,不良事件报告。FDA得出结论,大多数的评论,投诉,不良事件报告中提到的“服务”不足导致或促成不良事件和死亡,实际上与“再制造”有关。
就此,FDA发布了一份白皮书,公开了一份公众摘要,并举办了一次公共研讨会,以促进公众讨论服务和再制造之间的区别。白皮书描述了FDA对指导原则的最初想法,提供了一个流程图,并附有理解区别的文本,还包含了软件的补充方法、标签注意事项以及使用流程图的示例。FDA在白皮书中还提出了一些有针对性的问题,并寻求反馈。
FDA目前的想法:理解“再制造”和“维修”之间的区别很重要。
再制造是指对成品设备进行加工、调节、翻新、重新包装、恢复或任何其他行为,这些行为会显著改变成品设备的性能或安全规格或预期用途。FDA将原始设备制造商(OEM)合法销售的成品设备称为“合法销售的设备”。
维修是指成品设备中一个或多个零件在分销后的维修和/或预防性或日常维护,为了使其恢复到原始设备制造商制定的安全和性能规范,并满足其最初的预期用途。
无论实体的自认是“服务商”还是“再制造商”,FDA关注的是一个实体在特定设备上执行的具体活动。确定一个实体执行的活动是否为再制造影响到《食品和药品管理法》及其实施条例下监管要求的适用性和执行情况。FDA一直在执行FD&C法案及其实施条例对从事再制造的实体的要求,包括但不限于注册和上市、不良事件报告、质量体系(QS)条例和上市递交。
2、本指南草稿的重点
指南目的:
本指南草案涉及在拟重复使用和维护的设备上执行的活动。本指南草案讨论了原始设备制造商和第三方在此类设备上进行的活动是否可能是再制造。本指南草案的目的不是要采取重大的政策变化,而是要澄清FDA目前对适用定义的看法,并澄清(而不是改变)适用于再制造商的监管要求。
适用产品范围:
本指南范围内的产品是《食品、药品和化妆品法》第201(h)节中定义的设备,包括符合设备定义的软件和电子产品。一般来说,本指南中讨论的概念适用于所有可重复使用的设备,无论其分类为I类、II类或III类,包括需要上市前批准的设备。
本指南不适用于再加工的一次性设备。
重点定义:
服务(Service):在销售后,对成品器械中的一个或多个零件进行维修和/或预防性或例行维护,以使其恢复到原始设备制造商制定的安全和性能规范,并满足其原始的预期用途。维修不包括显著改变成品器械安全或性能规范或预期用途的活动。
再制造(Remanufacture):对成品器械进行加工(Process)、处理(condition)、翻新(renovate)、重新包装、恢复或任何其他行为,显著改变成品设备的性能或安全规格或预期用途。
修理(Repair):使部件恢复到原来规格的一种维修方式,包括在设备当前所有者的例行或定期保养之外更换非工作部件或零件。
修复/翻新/重建(Recondition/Refurbish/Rebuild):将医疗器械恢复到原始设备制造商的原始规格或“像新的一样”。如果对器械所做的更改没有显著改变成品器械的性能或安全规格或预期用途,则可以将器械恢复到当前规格。这些活动包括修理部件、安装OEM提供的更新和升级以及更换磨损部件。
如何判断所进行的活动是否是再制造?
四个指导原则:
1)评估是否会引起预期用途的变化?
2)确定是否单独或累积地显著改变成品器械的安全或性能规范?
3)评估器械的任何更改是否需要提交新的上市前递交?
4)评估是否会引起部件/零件/材料的尺寸和性能规格的变化,进而显著改变成品器械的安全或性能规范?
评估涉及部件/零件/材料的活动是否是再制造的流程图(不适用于软件):
FDA建议使用如下流程图来帮助确定其活动是否可能是再制造。
(提醒一下,这个流程图使用的前提:实体确定预期用途或设备灭菌方法、再处理指引、控制机制、工作原理或能源类型没有重大变化。
这个流程图仅作为视觉辅助工具,不包含所有相关考虑因素。使用流程图时,请参阅本节中的随附文本。)
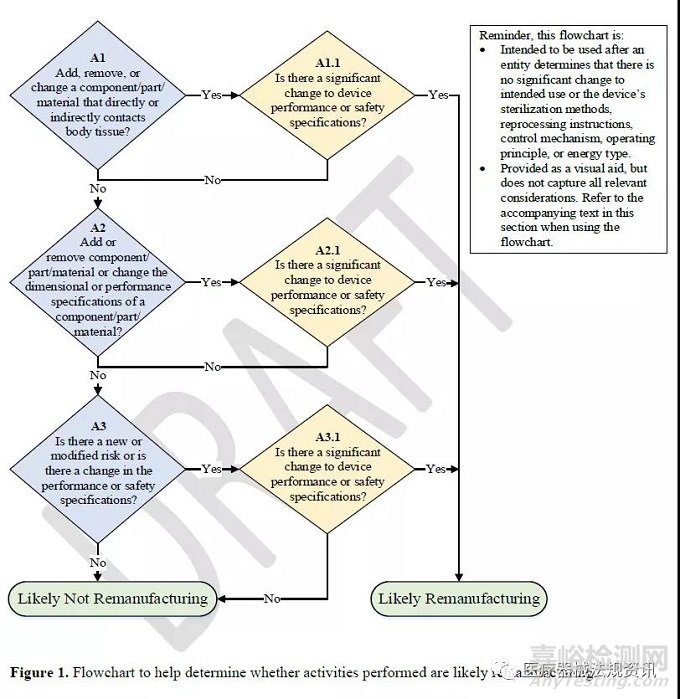
从以上流程图中可以看出,涉及部件/零件/材料的活动更有可能是再制造的因素包括以下:
1)添加、移除或更改直接或间接接触人体组织的器械部件/零件/材料导致器械性能或安全规范的重大更改;
2)添加或移除组件/零件/材料、或更改组件/零件/材料的尺寸或性能规格导致器械性能或安全规范的重大更改;
3)存在新的或修改过的风险,或性能或安全规范变化,导致器械性能或安全规范的重大更改。
哪些软件变更活动是再制造?
除了以下活动,其余软件变更的活动很有可能是再制造。
其余活动是否是再制造需要实体去评估是否导致器械性能或安全规范的重大更改,并保留这个评估记录。
代表OEM或OEM明确授权的活动,使合法销售的设备恢复其性能和安全规范以及预期用途;
执行OEM提供的更新和升级;
运行基于软件的硬件诊断;
评估病毒、恶意软件和其他网络安全相关问题;
重新安装OEM软件以恢复原始性能和安全规范;
将软件恢复到以前的配置;
安装OEM授权的网络安全更新;
打开或关闭与OEM预期用途一致的连接功能(如Wi-Fi和蓝牙连接);
执行数据备份和恢复操作;
评估软件库存;
收集系统日志;
管理用户帐户;和
访问诊断和维修信息。
是否是再制造的评估示例:

是否是再制造的评估文档示例:




来源:医疗器械法规资讯


