您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-12-17 21:28
医疗器械注册管理是医疗器械全生命周期管理中最为关键的环节之一,而在医疗器械注册进程中,又有三大关键环节,是需要大家注意的,它们将直接影响到你的注册提交以及成功审批的通过率:
1、非临床研究与产品检验
医疗器械非临床研究是指为评价医疗器械产品安全性和有效性,在实验室条件下对医疗器械产品进行的试验或者评价,主要包含:
(1) 产品化学和物理性能研究;
(2) 电气安全研究;
(3) 辐射安全研究;
(4) 软件研究;
(5) 生物学特性研究;
(6) 生物材料安全性研究;
(7) 消毒、灭菌工艺研究;
(8) 动物试验研究;
(9) 稳定性研究等等方面。
在该部分中,其基本要求包括:
非临床研究过程中确定的功能性、安全性指标及方法与产品预期使用条件、目的相适应,
研究样品具有代表性和典型性。
必要时,进行方法学验证、统计学分析。
非临床研究与产品检验是需要证据的,在申请注册/办理备案时,提交研制活动中产生的非临床证据,包括非临床研究报告综述、研究方案和研究报告等相关佐证材料。
详细的佐证材料要求如下:

2、临床评价
在临床评价方面,新办法与旧办法之间有十分明显的差异:
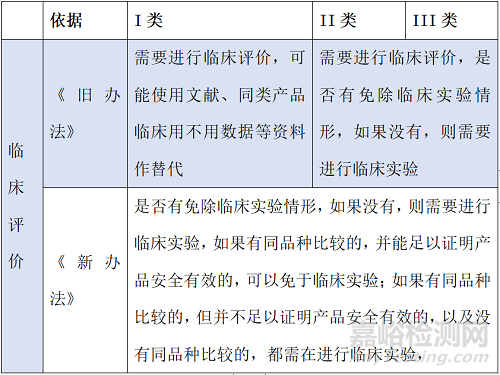
简单归类总结对比如下:

3、体系核查
在这部分,2020.03.10生效的《医疗器械注册质量管理体系核查指南》中对这部分有详细的规定要求:
首先需要确认的适用范围,对于第二类、第三类的医疗器械才需要医疗器械监管部门实施注册质量管理体系的现场核查。同时,对于在需要核查范围内的不同类不同生产地差异,按《医疗器械注册与备案管理办法》相关要求,其核查部门有所差异,具体来说,体现如下:
境内第二类:由申请人所在地省级药监部门组织开展;
境内第三类:由国家局器械审评中心通知申请人所在地的省级药监部门开展;
进口第二类、第三类:国家局器械审评中心认为有必要的,通知国家局审核查验中心开展。
现场质量管理体系核查按照医疗器械GMP要求开展,其重点核查的内容包括如下部分:
(1) 设计研发人员的资质
(2) 用于研发、样品试生产的厂房、设施、设备
(3) 文件管理(设计开发原始资料、受委托生产方文件等)
(4) 设计开发
(5) 采购(原辅材料、关键部件以及元器件)
(6) 生产(样品、生产管理体系)
(7) 质量控制(原材料、半成品、成品检验记录、项目规则)等。
当然,现场核查后会给出结论,一般有如下四种结果:
(1) 通过核查
(2) 整改后通过核查
(3) 未通过核查(不予以注册)
(4) 整改后未通过核查(不予以注册)
最后,祝每一次你提交的注册资料都顺利通过,现场核查都缺陷少少。都看到这里了,快关注我们吧。

来源:德大器械产业管家


