您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2023-07-06 20:03
|
报告基本情况 |
||||||
|
报告编码 |
系统自动生成 |
|||||
|
报告地 |
系统自动生成 |
|||||
|
1A报告人* |
报告表的填写人或报告的发起人 |
|||||
|
1.医疗器械情况 |
||||||
|
a 产品名称* |
输入产品注册证/备案凭证编号信息后会自动显示。 |
|||||
|
b 注册证编号*: |
如果注册证信息没有录入系统,将无法上报!如遇该情况,请联系注册人或所在区市场局系统管理员。 国食药监械(准/进)字xxxx第xxxxxxx号、国械注准/进xxxxxxxxxxx、国械注许xxxxxxxxxxx |
|||||
|
c 型号: |
|
|||||
|
d 规格: |
|
|||||
|
f 产品批号: |
无源产品和诊断试剂请提供批号。 |
|||||
|
g 产品编号: |
有源产品请提供编号或者序列号。 |
|||||
|
h UDI: |
|
|||||
|
i生产日期: |
|
|||||
|
j 有效期至: |
大型设备可以忽略此项,但耗材和诊断试剂请务必填写。 |
|||||
|
上市许可持有人名称 |
输入产品注册证/备案凭证编号信息后会自动显示。 |
|||||
|
2.不良事件情况 |
||||||
|
k 事件发生日期* |
如未知发生日期,可填写获知日期,并在s 使用过程中注明。 |
|||||
|
l 发现或获知日期* |
|
|||||
|
m 伤害* |
□死亡 □严重伤害 □其他(如可能导致死亡或严重伤害的医疗器械不良事件和/或故障) |
|||||
|
伤害表现:非必填项,部分产品数据库已开放,请尽可能在系统中选择。如无法选择,请用极简洁语言描述伤害表现。
|
||||||
|
n 器械故障表现:部分产品数据库已开放,请尽可能在系统中选择。如无法选择,请用极简洁语言描述器械故障,注意与伤害表现有所区分。
|
||||||
|
o姓 名: |
|
出生日期(选择) |
选择日期 |
|||
|
年龄类型: |
岁、月、天 |
年龄 |
填写数字 |
|||
|
既往病史 |
|
|||||
|
3.使用情况 |
||||||
|
p预期治疗疾病或作用: |
是指涉及的医疗器械用于治疗的疾病或作用。 |
|||||
|
q 器械使用日期*: |
|
|||||
|
r 使用场所*: |
□医疗机构 □家庭 □其他 |
场所名称: |
|
|||
|
s 使用过程*: |
对医疗器械实际使用过程的描述,按产品类别和实际情况尽可能提供,不超过 2000字。以下套用格式可以在填写时参考: 何时在何场所因何种原因开始使用何种医疗器械,使用情况如何,于何时出现何不良事件,给患者造成何种影响。何时采取何措施,何时不良事件表现治愈或好转。 |
|||||
|
t合并用药/械情况: |
与所报告器械配合使用的某品牌、规格、型号的其他器械或药品。 |
|||||
|
4.事件初步原因分析与处置仅限经营企业或使用单位填写 |
||||||
|
u事件原因分析*: |
□产品原因(包括说明书等)□操作原因□患者自身原因□无法确定 |
|||||
|
事件原因分析描述:
|
从产品质量、设计、运输保存、临床使用、患者疾病进展、合并用药械等方面分析。 |
|||||
|
初步处置情况*: |
对患者和/或对器械的处置情况。 |
|||||



|
报告基本情况 |
||||||
|
报告编码: |
系统自动生成 |
|||||
|
发生地: |
境内 |
|||||
|
1.医疗器械情况 |
||||||
|
a 产品名称* |
输入产品注册证/备案凭证编号信息后会自动显示。 |
|||||
|
b 注册证编号*: |
如果注册证信息没有录入系统,将无法上报!如遇该情况,请联系所在区市场局系统管理员。 |
|||||
|
曾用注册证编号 |
自动填充 |
|||||
|
曾用注册证编号上报*: |
□是 □否 |
|||||
|
c 型号: |
XXXX |
|||||
|
d 规格: |
|
|||||
|
e产地*: |
□进口 □国产 □港澳台 自动填充 |
|||||
|
产品类别*: |
□有源 □无源 □体外诊断试剂 自动填充 |
|||||
|
管理类别*: |
□III类 □II类 □I类 自动填充 |
|||||
|
f 产品批号: |
无源产品和诊断试剂请提供批号。 |
|||||
|
g 产品编号: |
有源产品请提供编号或者序列号。 |
|||||
|
h UDI: |
|
|||||
|
i生产日期: |
|
|||||
|
j 有效期至: |
大型设备可以忽略此项,但耗材和诊断试剂请务必填写。 |
|||||
|
注册人(上市许可持有人)名称 |
输入产品注册证/备案凭证编号信息后会自动显示。 |
|||||
|
2.不良事件情况 |
||||||
|
k 事件发生日期* |
如未知发生日期,可填写获知日期,并在s 使用过程中注明。 |
|||||
|
l 发现或获知日期* |
|
|||||
|
m 伤害* |
□死亡 □严重伤害 □其他(如濒临事件和/或故障) |
|||||
|
伤害表现:非必填项,部分产品数据库已开放,请尽可能在系统中选择。如无法选择,请用简洁语言描述伤害表现。
|
||||||
|
n 器械故障表现:部分产品数据库已开放,请尽可能在系统中选择。如无法选择,请用极简洁语言描述器械故障,注意与伤害表现有所区分。
|
||||||
|
o姓 名: |
|
出生日期(选择) |
|
|||
|
年龄类型: |
|
年龄 |
|
|||
|
性 别:□男 □女 |
病历号: |
|||||
|
既往病史: |
|
|||||
|
3.使用情况 |
||||||
|
p预期治疗疾病或作用: |
是指涉及的医疗器械用于治疗的疾病或作用。 |
|||||
|
q 器械使用日期*: |
|
|||||
|
r 使用场所*: |
□医疗机构 □家庭 □其他 |
场所名称: |
|
|||
|
s 使用过程*: |
对医疗器械实际使用过程的描述,按产品类别和实际情况尽可能提供,不超过2000字。以下套用格式可以在填写时参考: 何时在何场所因何种原因开始使用何种医疗器械,使用情况如何,于何时出现何不良事件,给患者造成何种影响。何时采取何措施,何时不良事件表现治愈或好转。 |
|||||
|
t合并用药/械情况: |
与所报告器械配合使用的某品牌、规格、型号的其他器械或药品。 |
|||||
|
4.事件调查 |
||||||
|
v是否展开了调查*: |
□是 □否 |
|||||
|
调查情况:
|
(可上传附件):调查情况可包含生产记录调查结果,医院使用情况,对退回的样品进行分析,抱怨趋势分析,设备维护保养状态等。 |
|||||
|
是否填写报告评价*: |
□是 □否 |
|||||
|
5.评价结果 |
||||||
|
w关联性评价*: |
□与产品有关□与产品无关□无法确定 |
|||||
|
x事件原因分析*: |
(可上传附件):需阐述是否涉及产品使用安全。 |
|||||
|
y是否需要开展产品风险评价* |
□是□否 选择是请填写“计划提交时间” 出现以下情况之一,注册人应进行产品风险评价: – 已知的不良事件,其发生概率超出预期; – 出现新的、未知不良事件; – 监管部门提示产品可能存在风险的; – 其它。 |
|||||
|
计划提交时间: |
|
|||||
|
6.控制措施 |
||||||
|
z是否采取了控制措施*: |
□是□否 |
|||||
|
具体控制措施描述: |
(可上传附件):为控制或降低系统性风险而对器械和/或配合使用单位采取的措施。 |
|||||
|
未采取控制措施原因: |
|
|||||
|
7.错报误报 |
||||||
|
是否错报误报*: |
□是 □否 若选择“是”,请填写错报误报原因 |
|||||
|
错报误报原因* |
|
|||||
|
8.报告合并 |
||||||
|
是否合并报告* |
□是 □否 若选择“是”,请填写合并报告编码 |
|||||
|
合并报告编码* |
|
|||||
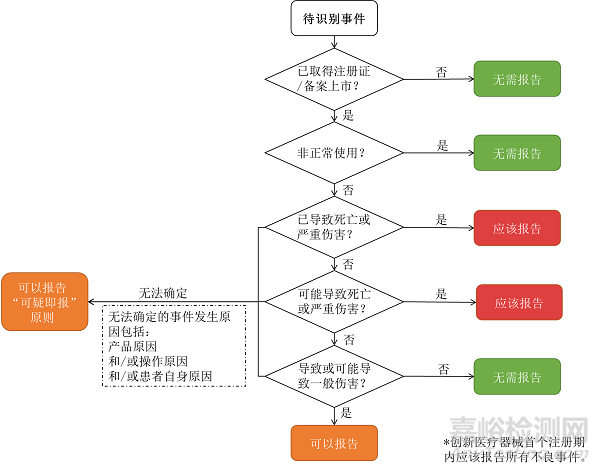
|
报告基本情况 |
||||||
|
报告编码: |
XXXXXXXXXXX |
|||||
|
报告日期: |
20XX年XX月XX日 |
|||||
|
报告人*: |
XXX |
|||||
|
单位名称: |
XXXX医院 |
|||||
|
联系地址: |
XXXXXXXXXXXX |
|||||
|
联系人: |
XXX |
|||||
|
联系电话: |
XXXXXXXX |
|||||
|
发生地: |
境内 |
|||||
|
1.医疗器械情况 |
||||||
|
a 产品名称* |
XXXXXXXXXX |
|||||
|
b 注册证编号*: |
国械注进XXXXXXXXXXX |
|||||
|
c 型号: |
XXXX |
|||||
|
d 规格: |
|
|||||
|
e产地*: |
■进口 □国产 □港澳台 |
|||||
|
产品类别*: |
■有源 □无源 □体外诊断试剂 |
|||||
|
管理类别*: |
■III类 □II类 □I类 |
|||||
|
f 产品批号: |
|
|||||
|
g 产品编号: |
XXXXX |
|||||
|
h UDI: |
|
|||||
|
i生产日期: |
20XX-XX-XX |
|||||
|
j 有效期至: |
20XX-XX-XX |
|||||
|
注册人(上市许可持有人)名称 |
XXXXXXXXXX公司 |
|||||
|
2.不良事件情况 |
||||||
|
k 事件发生日期* |
20XX-XX-XX |
|||||
|
l 发现或获知日期* |
20XX-XX-XX |
|||||
|
m 伤害* |
□死亡 □严重伤害 █其他 |
|||||
|
伤害表现:无 |
||||||
|
n 器械故障表现:治疗床自动升起。 |
||||||
|
o姓 名: |
XXX |
出生日期(选择) |
|
|||
|
年龄类型: |
|
年龄 |
XX |
|||
|
性 别:█男 □女 |
病历号: |
|||||
|
既往病史 |
|
|||||
|
3.使用情况 |
||||||
|
p预期治疗疾病或作用: |
医用直线加速器临床用于肿瘤的放射治疗。 |
|||||
|
q 器械使用日期*: |
20XX-XX-XX |
|||||
|
r 使用场所*: |
█医疗机构 □家庭 □其他 |
场所名称: |
XXXX医院 |
|||
|
s 使用过程*: |
XX医院于20XX年安装该设备,20XX年X月X日,当患者正接受治疗时,病床 自动升起并不断上升至患者碰到了设备上部的附件框,医护人员发现后立刻使用紧急按钮停止设备运作,随后关机并拆卸了电磁阀油压阀门,病床被卸油后落下。随后,医院对患者进行必要的检查,未发现患者因病床上升受到身体伤害。 |
|||||
|
t合并用药/械情况: |
无 |
|||||
|
4.事件初步原因分析与处置 仅限经营企业或使用单位填写 |
||||||
|
u事件原因分析*: |
█产品原因(包括说明书等)□操作原因 □患者自身原因□无法确定 |
|||||
|
事件原因分析描述:
|
现场维修工程师初步判定:床的基座由于雨水倒灌,造成短路,床因此自 动升起。 |
|||||
|
初步处置情况*: |
医护人员发现后立刻使用紧急按钮停止设备运作,随后关机并拆卸了电磁 阀油压阀门,病床被卸油后落下。随后,医院对患者进行必要的检查,未 发现患者因病床上升受到身体伤害。 |
|||||
注册人(上市许可持有人)医疗器械不良事件报告表填写案例
|
报告基本情况 |
||||||
|
报告编码: |
XXXXXXXXXXX |
|||||
|
报告日期: |
20XX年XX月XX日 |
|||||
|
报告人*: |
XXX |
|||||
|
单位名称: |
XXXX医院 |
|||||
|
联系地址: |
XXXXXXXXXXXX |
|||||
|
联系人: |
XXX |
|||||
|
联系电话: |
XXXXXXXX |
|||||
|
发生地: |
境内 |
|||||
|
1.医疗器械情况 |
||||||
|
a 产品名称* |
XXXXXXXXXX |
|||||
|
b 注册证编号*: |
国械注进XXXXXXXXXXX |
|||||
|
c 型号: |
XXXX |
|||||
|
d 规格: |
|
|||||
|
e产地*: |
■进口 □国产 □港澳台 |
|||||
|
产品类别*: |
■有源 □无源 □体外诊断试剂 |
|||||
|
管理类别*: |
■III类 □II类 □I类 |
|||||
|
f 产品批号: |
|
|||||
|
g 产品编号: |
XXXXX |
|||||
|
h UDI: |
|
|||||
|
i生产日期: |
20XX-XX-XX |
|||||
|
j 有效期至: |
20XX-XX-XX |
|||||
|
注册人(上市许可持有人)名称 |
XXXXXXXXXX公司 |
|||||
|
2.不良事件情况 |
||||||
|
k 事件发生日期* |
20XX-XX-XX |
|||||
|
l 发现或获知日期* |
20XX-XX-XX |
|||||
|
m 伤害* |
□死亡 □严重伤害 █其他 |
|||||
|
伤害表现:无 |
||||||
|
n 器械故障表现:治疗床自动升起。 |
||||||
|
o姓 名: |
XXX |
出生日期(选择) |
|
|||
|
年龄类型: |
|
年龄 |
XX |
|||
|
性 别:█男 □女 |
病历号: |
|||||
|
既往病史 |
|
|||||
|
3.使用情况 |
||||||
|
p预期治疗疾病或作用: |
医用直线加速器临床用于肿瘤的放射治疗。 |
|||||
|
q 器械使用日期*: |
20XX-XX-XX |
|||||
|
r 使用场所*: |
█医疗机构 □家庭 □其他 |
场所名称: |
XXXX医院 |
|||
|
s 使用过程*: |
XX医院于20XX年安装该设备,20XX年X月X日,当患者正接受治疗时,病床 自动升起并不断上升至患者碰到了设备上部的附件框,医护人员发现后立刻使用紧急按钮停止设备运作,随后关机并拆卸了电磁阀油压阀门,病床被卸油后落下。随后,医院对患者进行必要的检查,未发现患者因病床上升受到身体伤害。 |
|||||
|
t合并用药/械情况: |
无 |
|||||
|
4.事件调查 |
||||||
|
v是否展开了调查*: |
█是 □否 |
|||||
|
调查情况:
|
(可上传附件): 经XX公司专家团队调查后发现: XX医院于20XX年安装和调试该设备,XX公司末次现场维修时间为20XX年XX月XX日。 本次事件发生后,医院对患者进行必要的检查,未发现患者因病床上升受到身体伤害。 XX公司已组织技术组对该事件进行调查分析,结论为该事件系床的基座浸入雨水造成短路所致。 截止到目前为止,在中国境内的同类产品未曾发生过类似事件。 XX公司将发布一封客户告知信(内部编号为:XXXX-XX-XX),以告知中国客户将对该台设备安装浮止装置,如果安装了浮止装置,即使雨水浸入基座,浮止装置一旦漂浮起来,则设备会自动断电。 XX公司将准备一项现场纠正措施(内部编号为:XXXX-XX-XX)对该台设备安装浮止装置以解决此潜在问题。 XX公司将近期为该台设备补装浮止装置。 |
|||||
|
是否填写报告评价*: |
█是 □否 |
|||||
|
5.评价结果 |
||||||
|
w关联性评价*: |
█与产品有关□与产品无关□无法确定 |
|||||
|
x事件原因分析*: |
(可上传附件):需阐述是否涉及产品使用安全。 |
|||||
|
y是否需要开展产品风险评价* |
█是□否 以下风险评价报告只是演示样例,实际应在系统的“产品风险评价报告”模块中提交: XX公司已组织技术组对该事件进行调查分析,结论为该事件系床的基座浸入雨水造成短路所致。 风险评价简述: 1. 由于基建要求系统安装必须处于室内,并由医院施工团队验收,遭受雨水渗透可能性较低。 2. 截止到目前为止,在中国境内的同类的未曾发生过类似事件。 3. 根据全球装机量及使用情况,结合同样原因导致的雨水浸入设备情况数据可得出:该事件发生的概率较低。 4. 操作者并不被鼓励在室内环境有雨水的情况下使用仪器,以防触电危害的发生。 基于以上的风险分析,风险评估结果为XX(风险严重程度为X,发生概率为X)。 |
|||||
|
计划提交时间: |
20XX-XX-XX |
|||||
|
6.控制措施 |
||||||
|
z是否采取了控制措施*: |
█是□否 |
|||||
|
具体控制措施描述: |
(可上传附件):为控制或降低系统性风险而对器械和/或配合使用单位采取的措施。 |
|||||
|
未采取控制措施原因: |
|
|||||
|
7.错报误报 |
||||||
|
是否错报误报*: |
□是█否 |
|||||
|
8.报告合并 |
||||||
|
是否合并报告* |
□是█否 |
|||||

来源:上海市药品监督管理局