您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-07-02 09:58
医用超声雾化器、医用压缩式雾化器是以超声振荡或气体压缩机驱动的方式,将药液转化为气雾颗粒的有源医疗器械,预期用于对液态药物进行雾化,供患者吸入治疗使用。
按照《医疗器械分类目录》,该类产品属于08-05-07雾化设备/雾化装置,管理类别为第二类。
一、医用雾化器的结构组成和工作原理
1.结构及组成
根据产品实际情况描述产品的结构及组成,不使用“主要”“等”字进行描述。产品结构及组成可能的情况如下:
1.1医用超声雾化器一般由主机、雾化杯、连接管、咬嘴或吸入面罩组成,其中的主机一般由超声波发生器(超声换能器)、超声薄膜、送风装置、调节和控制系统组成。
1.2医用压缩式雾化器一般由主机、连接管(送气管)、雾化装置、咬嘴或吸入面罩组成,其中主机一般由压缩泵、过滤组件和控制系统组成。
1.3产品结构和组成描述的示例
医用超声雾化器产品实例如图1所示。
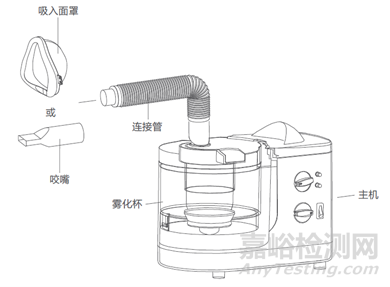
图1 医用超声雾化器产品示例
医用压缩式雾化器产品实例如图2所示。
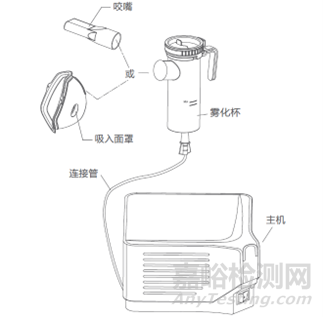
图2 医用压缩式雾化器产品示例
2.工作原理
2.1医用超声雾化器工作原理
超声雾化器由超声波发生器产生的高频电流经过安装在雾化缸里的超声换能器使其将高频电流转换为相同频率的声波,由换能器产生的超声波通过雾化缸中的耦合作用,通过雾化杯底部的超声薄膜,从而使超声波直接作用于雾化杯中的液体。当超声波从杯底经传导到达药液表面时,液—气分界面即药液与空气交界处,在受到垂直于分界面的超声波的作用后(即能量作用),使药液表面形成张力波,随着表面张力波能量的增强,当表面张力波能量达到一定值时,在药液表面的张力波波峰也同时增大,使其波峰处的液体雾粒飞出(雾粒直径的大小随超声波的频率增大而缩小)。由于超声波产生的雾粒具有尺寸均一,动量极小,故容易随气流行走,药液产生雾粒的数量随超声波能量的增加而增多(即超声波的功率越大,则产生的雾粒的数量越多)。在医用超声雾化器将药液分裂成微粒后,再由送风装置产生的气流作用而生成药雾,药雾经连接管输送给患者,如图3、图4所示。
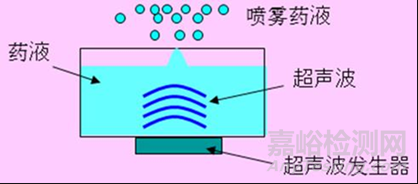
图3 医用超声雾化器雾化装置示例图
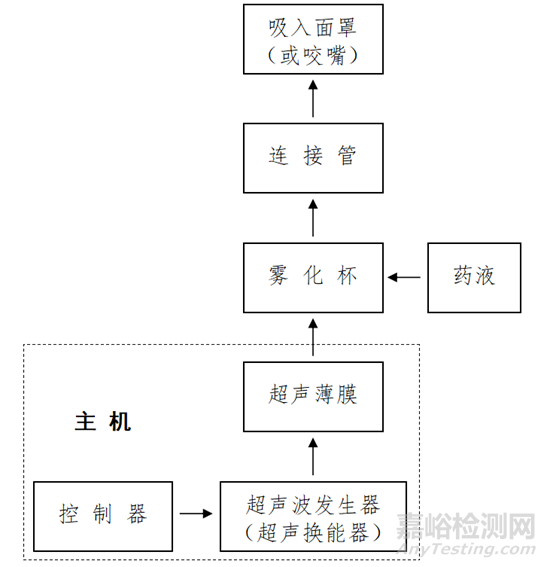
图4 医用超声雾化器工作原理示意图
2.2医用压缩式雾化器工作原理
医用压缩式雾化器应用的是文丘里效应原理,一般是通过气体压缩机产生的压缩气体为驱动源来产生及传输气雾的,其工作原理示意图如图5所示,其中的雾化装置工作原理示例如图6所示:压缩机产生的压缩空气从喷嘴喷出时,通过喷嘴与吸水管之间产生的负压作用,向上吸起药液。吸上来的药液冲击到上方的隔片,变成极细的雾状向外部喷出,如图7所示。

图5 医用压缩式雾化器工作原理示意图
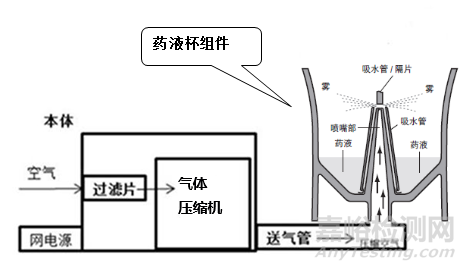
图6 医用压缩式雾化器工作原理示例图
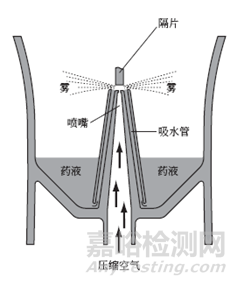
图7 医用压缩式雾化器雾化装置图示例
二、医用雾化器的主要风险
医用雾化器的主要危险(源)(举例)见表
表 产品主要危险(源)(举例)
|
危险(源)的分类 |
危险(源)的形成因素 |
可能的后果 |
|
|---|---|---|---|
|
能量风险 |
气流压力 热量 |
能量危害、操作危害、 电气故障、过热。 |
连接管在治疗中拔下,由于气压的作用使用户受伤。烫伤,烟雾。 |
|
电磁兼容风险 |
电磁能危害 |
由于主机动作产生的电磁波的影响。 |
引起其他医疗机器的误动作。 |
|
机械风险 |
噪音 |
长时间使用。 |
长时间置于噪音环境中的危害。 |
|
生物学危害 |
生物污染 |
生产环境控制不好。 灭菌操作不严格、包装破损。 微生物限度要求产品微生物指标不符合要求。 |
产品带菌或微生物限度指标不符合要求,引起患者呼吸道感染。 |
|
生物不相容性 |
残留物过多。 添加其他化学物质。 |
PVC:氯乙烷超标、增塑剂量过大,产生毒性或刺激;邻苯二甲酸二酯(DEHP)含量大。 |
|
|
不正确的配方 (化学成分) |
未按照工艺要求配料。 添加剂或助剂使用比例不正确。 |
有可能引起小分子物质残留量过大,造成毒性危害。 |
|
|
生物学危害 |
毒性 |
不正确的配方、添加。 加工工艺控制不严格。 后处理工艺控制不严格。如:环己酮用量较多或环己酮配方毒性大。 |
生物相容性不符合要求。 |
|
再感染和/或交叉感染 |
使用不当、标识不清。 |
引起感染、交叉感染。 |
|
|
环境 危害 |
储存或运行偏离预定的环境条件 |
储运条件(如温度、湿度)不符合要求。 |
产品老化, 无菌有效期缩短。 |
|
意外的机械破坏 |
储运、使用过程中发生意外的机械性破坏。 |
产品使用性能无法得到保证。 |
|
|
由于废物和(或)医疗器械处置的污染 |
使用后的产品没有按照要求集中销毁。 |
造成环境污染或细菌的交叉感染。 |
|
|
与医 疗器 械使 用有 关的 危害 |
不适当的标记 |
标记不清晰、错误, 没有按照要求进行标记。 |
错误使用、 储存错误、 产品辨别错误。 |
|
不适当的操作说明,如: (1)和医疗器械一起使用的附件规范不适当; (2)预先检查规范不适当; (3)操作说明书不准确、清晰。 |
包装破损无法识别。 操作要点不突出。
|
无法保证使用安全性。 导致操作失误。
|
|
|
与医 疗器 械使 用有 关的 危害 |
由不熟练/未经培训的人员使用 |
操作不熟练、操作失误。 规格型号选用错误、成人小儿或新生儿混用。 连接不正确或不到位。 |
导致雾化气体未能输入呼吸系统。 吸入雾化气体效率降低、鼻孔或耳挂固定处皮肤损伤。 雾化气体泄漏。 |
|
对副作用的警告不充分 |
对操作人员警示不足。 |
重复使用。 二次灭菌。 已过期的产品被使用。 |
|
|
对一次性使用医疗器械很可能再次使用的危害警告不适当 |
造成重复使用。 |
交叉感染。 |
|
|
不适当不合适或过于复杂的使用者接口 |
违反或缩减说明书、程序等 |
操作方法、注意事项、储存方法、警示事项等表述不清。 |
不能实现预期雾化气体吸入功能、重复使用引起感染、没有集中销毁造成环境危害等。 |
|
功能性失效和老化引起的危害 |
对医疗器械寿命终止缺少适当的决定 |
没有标识产品有效期。 |
超出有效期的产品被使用,造成细菌感染或因材料老化产生而导致产品性能不符合要求(如输氧管破损、连接松动等)。 |
|
不适当的包装(医疗器械的污染和/或变质) |
没有进行包装确认。 |
不能确保产品无菌,从而导致出现细菌感染。 |
|
|
再次使用和/或不适当的再次使用 |
产品标识没有明确。 |
出现细菌感染、交叉感染、管路老化破损。 |
|
三、医用雾化器性能研究实验要求
1、检测方法研究
1.1应明确进行产品技术要求中标称雾化速率(或喷雾速率)、雾粒空气动力学特性等研究所用溶液/混悬液的成分、浓度。所用溶液/混悬液成分、浓度不同,对结果均会造成影响。建议采用相关标准(YY/T 1743《麻醉和呼吸设备雾化系统和组件》)中气雾颗粒输出、喷雾速率和颗粒直径的测试方法对所述溶液进行研究。
1.2应明确产品技术要求中所载明的无菌检测方法或微生物限度及控制菌检测方法的确定依据,如无菌检测按照中华人民共和国药典2020年版四部1101无菌检查法进行,微生物限度检测按照1105微生物限度检查法进行,控制菌按照1106控制菌检查法进行;需要根据样品的特点,建立并验证样品的微生物检测方法并验证。
1.3应对产品技术要求中所载明化学检测浸提液的制备方法进行研究与验证,是否参照相关标准(GB/T 14233.1 《医用输液、输血、注射器具检验方法第1部分:化学分析方法》)中检验液制备所述方法进行浸提液的制备;是否对环氧乙烷检测的样品制备及检测方法进行研究并验证;是否对化学指标的值进行研究,明确制定的依据并进行验证。
2、生物学特性研究
应明确产品直接或间接与患者接触部件的材料及材料表征。参照《关于印发医疗器械生物学评价和审评指南的通知》及相关标准(GB/T 16886.1《医疗器械生物学评价第1部分:风险管理过程中的评价与试验》)的要求,根据接触条件、接触性质、程度、频次和时间明确器械的类别及评价路径,对其进行生物相容性评价。生物相容性研究原则上不用原材料代替成品,若使用原材料代替成品进行生物相容性试验,宜对原材料使用与成品相同的生产制造工艺及微生物控制方式处理后进行生物相容性试验。
建议根据YY/T1778.1(ISO 18562)考虑气体通路相容性要求。
3、灭菌/消毒工艺研究
3.1对于出厂时已进行微生物控制且在初次使用前不需使用者进行微生物控制的产品组件,若其为无菌提供,则应经过一个经确认的过程使产品无菌,明确无菌保证水平(SAL)并开展灭菌过程的确认;若其非无菌提供,应明确微生物控制方法(工艺过程方法和控制参数),可参考相关标准(GB 15982 《医院消毒卫生标准》对中度危险性医疗器材的要求,确定微生物限度的性能指标,并可参照《中华人民共和国药典》2020年版四部,明确对控制菌的要求。
3.2若产品组件非一次性使用(一般包含出厂时已进行微生物控制的与未进行微生物控制的),则需要使用者在使用前或使用过程中进行清洗消毒;若产品组件一次性使用但出厂前未进行微生物控制,则需要使用者在使用前进行消毒。
应当明确推荐的清洗消毒工艺(方法和参数),以及所推荐的清洗消毒方法确定的依据,并开展清洗消毒效果的验证。需要由使用者进行消毒的部件一般包括与患者直接接触的吸入装置、延长装置、药杯及主机等。
3.3残留毒性研究
若所使用的微生物控制方法容易出现残留,如环氧乙烷灭菌或消毒,应明确残留物的信息及采取的处理工艺,并开展验证。若采用多种处理工艺,则应分别开展验证研究。
4、产品有效期和包装研究
4.1参照《有源医疗器械使用期限注册技术审查指导原则》的要求进行使用期限的研究.
4.2对产品有效期的确定可以分为主机与吸入附件两部分,分别对这两部分进行研究,以确定产品的使用期限及货架有效期。
若吸入附件经过微生物控制后出厂,应开展验证明确其灭菌/消毒有效期。若吸入附件及液体容器可重复使用,则需对其使用次数/期限进行验证,并开展验证,验证过程中需要关注清洗、消毒、拆卸、组装方式对使用次数/期限的影响。
4.3包装及包装的完整性
出厂前已进行微生物控制的产品组成部分的包装,应明确与产品直接接触的内包装材料,以及与微生物控制手段的适应性,无菌提供的应明确内包装初始污染菌的可接受程度。可参考相关标准(YY/T 0681《无菌医疗器械包装试验方法》系列标准)对包装性能进行研究,并开展验证,验证内容至少包含包装的密封性能、剥离强度等指标。
对最小销售单元包装及运输包装进行跌落、堆码、运输等试验,以验证包装对贮存、运输中环境条件(例如:震动、振动、温度和湿度的波动)的适应性。
4.4环境试验要求
医用电器环境要求是评价产品在各种工作环境和模拟贮存、运输环境下的适应性。可按照GB/T14710制定不同的气候环境条件和机械环境条件来进行试验,或通过对关键部件的试验来评价整机的情况,也可以通过已上市同类产品比对方式进行判断。
5、软件研究
雾化器所含医用软件一般为软件组件,参照《医疗器械软件注册审查指导原则》(2022修订版)的相关要求,开展软件研究。
如适用,参照《医疗器械网络安全注册审查指导原则》(2022修订版)的要求开展相应研究。
如果产品采用无创“移动计算终端”实现一项或多项医疗用途,如使用蓝牙与智能手机连接并由智能手机对雾化器进行控制的产品,参照《移动医疗器械注册技术审查指导原则》的要求开展相应的研究。
6、内部电源供电的,开展使用可充电内部电源充满电后可工作的最长时间或可进行雾化治疗的次数的研究。
7、开展液体容器、雾化装置与药液的相容性研究。
8、雾粒空气动力学特性研究
开展典型药物雾粒空气动学研究。若说明书中宣称用于某一种或某一类药物的雾化治疗,开展该药物雾粒空气动力学特性(MMAD、气雾颗粒输出、喷雾速率)研究。
9、开展使用混悬液或高粘度药液、温度敏感等药物超声雾化适用性的研究(若适用)。

来源:嘉峪检测网


