您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-01-28 22:31
本文整理了做医疗器械CE认证时详细的检测和验证内容。
01、性 能 检 测
1、产品性能检测
依据产品标准或者技术要求,对产品进行的全性能检测。
接骨板:材料,机械性能,表面质量,尺寸,静态四点弯曲,疲劳四点弯曲。
接骨螺钉:材料,机械性能,表面质量,尺寸,螺钉旋入旋出,扭转,自攻,静动态四点弯曲。
2、选样说明
一般选择最具代表性,最差情况型号进行检测。如果一个型号无法覆盖,需测试2个或者多个型号。
3、测试机构
如果企业有设备有能力检测,可以企业自测。否则就找有资质的第三方测试。
02、生 物 相 容 性
1、生物相容性检测
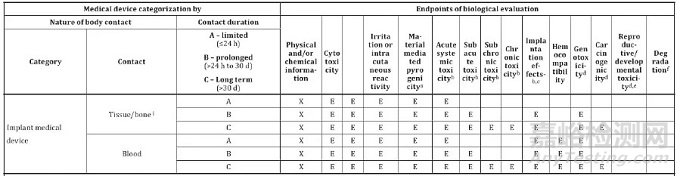
根据ISO10993-1:2018 附录A,骨科植入物需要检测的项目有:细胞毒,致敏,刺激,热原,急性全身毒性,亚急性毒性,亚慢性毒性(15/30周),慢性毒性,植入反应(30/60周),遗传毒性,致癌性。
(检测成品)
评估可以包括对相关的现有临床前和临床数据以及实际测试的回顾。这样的评估可能得出这样的结论:如果该材料具有可证明的安全使用历史,并且具有与设计中的医疗设备相同的指定作用和物理形式,则无需进行测试。可以证明等效性的信息类型包括在附录B中。当已经有足够的信息来对材料和/或医疗器械进行风险评估时,通常不必进行测试。
03、特 殊 过 程 验 证
特殊过程:以下情况之一的,可以认定为特殊工序或过程:
1、不能通过后续的监视和测量加以验证,或即使能够验证,但产生的不合格不能纠正的过程;
2、过程的产品不能快速、经济的直接进行验证,或需要经破坏性试验或采用较复杂或成本较高、试验周期较长的方法才能测量,或只能通过间接的监控的过程;
3、测过程的产品只有在产品使用或交付后,不合格的质量特性才暴露出来的过程
生产过程中特殊过程有内包装封口验证,灭菌验证,清洗验证等。
封口验证
即对封口参数(温度,压力,时间/速度)的验证。
灭菌验证
如果产品为无菌提供,需对整个灭菌过程进行验证;如果产品为非无菌提供,由医院使用前灭菌,则说明书中需提供已验证的灭菌参数。
无菌提供产品:生产商需对整个灭菌过程进行验证,包括IQ, OQ和PQ。
EO灭菌:EN ISO 11135: 2014
辐射灭菌:EN ISO 11137-1: 2015/A2:2019, EN ISO 11137-2: 2015
包括剂量设定和剂量分布,确定最大辐照剂量,有效期验证的样品应先用最大剂量辐照。生物负载监测,剂量审核。
湿热灭菌:EN ISO 17665-1:2006,EN ISO 17665-2:2009
干热灭菌:ISO 20857:2010
需定期进行再验证,通常是每年要再验证。产品的变更、灭菌设施和工艺的变更、其他可能对验证造成影响的情况也需进行再验证。
如企业自己灭菌,相关检测,验证和再验证将是审核重点。如委外灭菌需要选择有资质的灭菌站,签订灭菌协议。也需按照体系要求对灭菌公司进行供应商审核。
灭菌控制程序:灭菌验证,常规灭菌控制,产品放行,再验证
非无菌提供使用前灭菌的产品:需对说明书中列明的灭菌参数进行验证。注意:应该使用欧洲常用的灭菌参数。
鉴于对蒸汽灭菌的成熟认识,标准以及实践已经不再要求做微生物测试,只需要通过物理方法证明产品内外表面都能够达到灭菌条件即可。验证过程需做三次循环。
接收标准:以预真空 134 ℃, 3min 蒸汽灭菌为例
◆ 灭菌温度范围:134~137 ℃
◆ 中小型灭菌器平衡时间应小于15s,大型灭菌器应小于30s。
◆ 所有温度测量点都应在134~137℃温度范围内,并且不同点间最大温度差不大于2℃。
◆ 全周期中灭菌保持时间要不小于3min。
清洗验证
包括生产过程的清洗验证和医院使用前的清洗消毒验证。
生产过程的清洗验证: 骨科植入物验证依据 ISO 19227,接收标准包括:目视检查,生物负载,微粒污染,细胞内毒性,有机污染物,无机污染物,细胞毒性。
选样说明:选择结构复杂,较难清洗的产品为代表产品。
清洗参数:水温,水量,清洗时间,是否添加清洗剂,烘干温度,烘干时间。
使用前的清洗消毒验证:验证依据:ISO 17664, ISO 15883-1, -2, -5. 接收标准:
1.检测液中无可视残留物。
2.清洗后的器械上最大蛋白质含量应低于下面表格中的行动线要求。

04、包 装 及 有 效 期 验 证
包装验证即无菌屏障系统验证:依据ISO11607-1/-2, 验证项目:
1、包装材料与成型和密封过程的适应性;
2、包装材料与预期灭菌过程的适应性;
3、包装材料与标签系统的适应性;
4、包装材料与贮存运输过程的适应性;
5、包装材料的微生物屏障特性;
6、包装材料的生物相容性特性;
7、包装材料的物理化学性能;
注意:无菌产品或者使用前灭菌的产品的内包材需在洁净室生产,需要供应商提供洁净室检测报告。
有效期验证:包括包装有效期和产品有效期,因此需同时验证包装和产品的有效期。可以实时老化和加速老化。
05、关 键 工 序 验 证
关键工序:一般来说,以下情况之一的,应该被认定为关键工序:
1、对产品质量、特性、功能、寿命、可靠性及成本等有直接影响的工序;
2、产品重要或关键特性形成的工序;
3、工艺复杂、质量容易波动、对人员技能、设施能力、环境条件要求较高,或以往生产过程中发生问题较多的工序;
4、若控制不好,可能存在产品质量隐患的工序;
5、存在环境污染和人员职业健康安全隐患或风险的工序。
所有特殊过程和关键工序的验证,都必须有详细的验证方案和验证报告,并且验证报告中需附上原始记录。再验证要求和频次,需在验证方案和报告中写明。

来源:微珂器械服务


