您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-06-16 09:40
本文主要针对高效液相色谱法测定有关物质的方法学验证。任何技术贴都不可避免存在不足和争议,我们也在行进中不断进步和纠偏,未尽之处欢迎讨论与沟通。
一、方法学验证汇总表
先来一个张表格,送给各位读者,这个表格既是有关物质方法学验证试验中的标杆和尺度,也作为申报资料中的一部分。现将梗概罗列汇总如下,坐好别抢,见者有份:
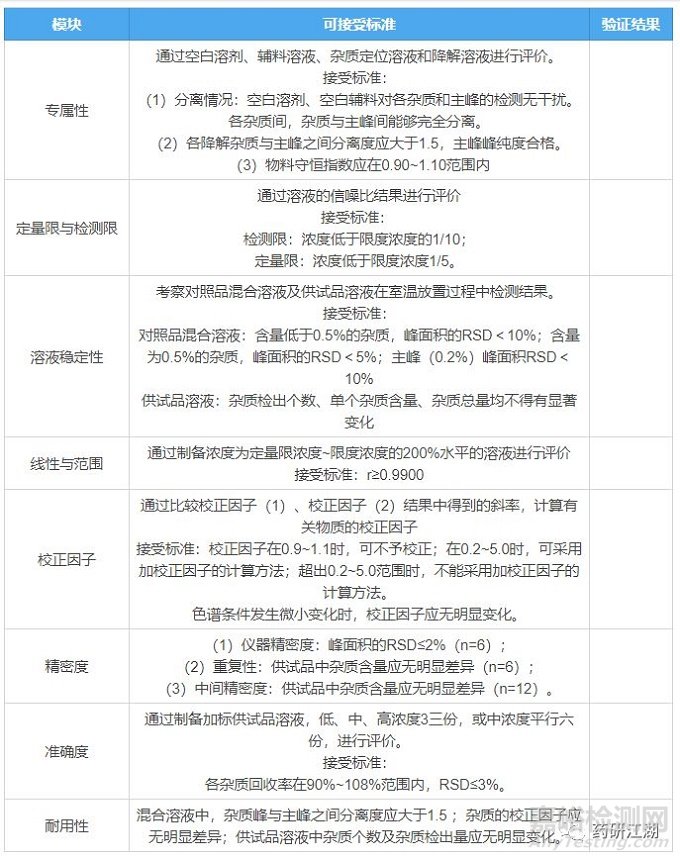
二、方法学验证内容
这个版块海棠君将汇总表格中各项试验模块具体步骤和注意事项掰开详述。有且不仅有这些内容,研发品种千差万别,请根据自身情况做相应调整。
1.专属性试验
有关物质的专属性指在其他成分(如杂质、降解产物、辅料组分等)可能存在的情况下,采用的分析方法能够准确测定出待测杂质的特性,应包括杂质分离度考察试验和强制降解试验两部分。
专属性是方法学验证的前提。若方法不专属,后续一切验证都是徒劳。
1.1杂质分离度考察
空白溶剂:按质量标准规定配制。
空白辅料溶液:按处方比例配制并混匀,按质量标准中供试品的配制方法配制。
各定位溶液:应包括所有研究的杂质和主成分。各定位溶液的浓度应适宜,太小则不易辨认,太大则峰太宽不易定位保留时间。
供试品溶液:按质量标准规定配制。
混合溶液:其中组分包括主成分(测定浓度)和各已知杂质(不得小于限度浓度)。
空白溶剂和空白辅料若有色谱峰,则应在质量标准中写明扣除。
1.2强制降解试验
强制降解试验旨在考察色谱条件对降解产物与各已知组分之间的分离能力、样品可能存在的降解途径,也可大概推测样品的稳定性情况(由于强制降解试验条件过于剧烈,稳定性情况未必准确)。
破坏条件一般包括:酸、碱、氧化、高温、光照,有时也根据需要进行高湿或者高温高湿的条件考察。采用上述条件降解样品,研究可能存在的降解产物和降解途径对杂质测定的影响,并对主成分峰进行峰纯度检查。
配制样品:原料药、辅料、自制品、市售品。有关物质的强制降解试验要考察降解杂质的来源,不应忽略空白辅料的配制。每个序列中推荐分析含有各已知杂质的混合溶液,作为供试品溶液所检出杂质的归属依据。
注意事项:
a.降解条件要适度,以主成分峰面积降解至80%~90%为宜。
b.为避免有小极性的物质降解产生,没有被充分洗脱出来,应视情况将采集时间延长。
c.计算质量守恒指数时,应考虑到杂质的校正因子。
d.将溶液放置于高温条件下的时候,应置于水浴锅中,而不是放在烘箱中。
e.为避免溶剂峰过大而影响杂质检出,酸、碱、和氧化所用试剂使用量在可浸润样品的情况下尽量少。
试验结果给出后,我们应思考和注意这些问题:
a.当原料药、自制品、市售品的降解趋势不同时,应分析并说明原因。
b.若有较大未知杂质降解产生,应对其进行界定,并分析是否应对该杂质进行研究。
c.与含量测定方法学验证中的降解试验结果进行对比,分析降解程度是否具备合理性。
e.强制降解试验的结果也许不能被准确重复出来,这是正常的,但应控制试验条件和试验环境,尽量避免试验结果的偶然性。
2.定量限与检测限
定义:
定量限系指待测组分能够被准确定量测定的最低量。
检测限系指待测组分能够被可靠地检测出来的最低量。
二者能够体现出色谱系统检测有关物质的灵敏度情况。
一般用信噪比作为参数来呈现灵敏度。信噪比即把已知低浓度试样测出的信号与噪声信号进行比较,用S/N表示。一般情况下,将信噪比约为3:1时相应浓度或注入仪器的量确定检测限,将信噪比约为10:1时相应浓度或注入仪器的量确定定量限。
配制溶液时,可先配制限度浓度的混合溶液,得出各组分的信噪比,再根据信噪比逐级稀释至S/N=10或3。有时为方便操作,也可使用线性溶液进行逐级稀释。
可以混合进样,也可以单个或部分混合进样。没有硬性要求。
注意:为确保检测方法的灵敏度,推荐考察不同仪器和试验条件下的定量限与检测限情况。
3.溶液稳定性
旨在考察对照品溶液/自身对照溶液、供试品溶液、系统适用性溶液在一定检测时间内的稳定性情况。
对照品溶液/自身对照溶液以峰面积变化为指标考察稳定性。
供试品溶液以杂质检出个数、单个杂质含量、杂质总量的变化为指标考察溶液稳定性。
系统适用性溶液按质量标准规定需符合要求,并不应有较大降解峰产生。当配制系统适用性溶液所需已知杂质对照品不易获得时,也推荐考察放置较长时间的稳定性,以便配制后保存留用,无需每次试验配制系统适用性溶液。
对试验结果的思考:
a.若样品对热不稳定,可考察样品盘冷藏控温放置,冷藏过程中注意观察样品在低温下是否有析出现象。
b.若样品对光照不稳定,可考察避光放置或棕色瓶放置。
c.根据化合物的化学结构和特性,有时换用溶剂可以改善溶液稳定性。
d.溶液稳定性结果是今后样品检验时间的依据,不稳定的样品应在质量标准中写明临用现配。
4.线性与范围
旨在验证在各杂质的测定范围内,待测组分响应值与浓度的线性关系。
制备一个高浓度的对照品贮备液,逐步稀释一系列不同浓度的对照品溶液。各杂质浓度推荐范围为定量限~限度浓度的200%,分别进样分析。以浓度为横坐标,以峰面积为纵坐标,进行线性回归,绘制线形图,报告回归方程和相关系数。
注意:
a.各杂质限度浓度的20%应至少大于定量限浓度。
b.线性溶液稀释时,应考虑移取体积的合理性。
c.制备至少5个以上浓度点,尽量均匀分布。
5.校正因子
采用主成分对照品和杂质对照品的标准曲线斜率比值进行比较获得。
当质量标准中规定了杂质的校正因子时,应对校正因子进行全面的验证,确保数值准确。
包括准确度(用加校正因子方法计算回收率)、精密度(采用不同仪器、由不同人员考察)、耐用性(当色谱条件发生微小变化时,校正因子不应变化)等验证内容。
校正因子在质量标准中的规定:当校正因子在0.9~1.1时,可不予校正;0.2~5.0时,可采用加校正因子的计算方法;超出0.2~5.0范围时,不能采用加校正因子的方法计算。
6.精密度
6.1仪器精密度
目的:为保证仪器每次所测对照品溶液/自身对照溶液峰面积均一致,以确保有关物质结果的准确性,设计仪器精密度试验。
取对照品溶液/自身对照溶液,连续进样6次。考察保留时间和峰面积的RSD%。
6.2重复性
目的:在相同条件下,由同一个分析人员测定结果的精密度。
1.按照拟定标准配制空白溶剂、对照品溶液/自身对照溶液和6份供试品溶液,计算6份供试品溶液的含量,计算平均值并统计6份结果的RSD值。
2.可用加标法进行考察,具体可参考准确度试验项下。
2015年版中国药典关于重复性的可接受范围截图如下:

6.3中间精密度
目的:考察在同一实验室,不同时间,由不同人员用不同设备测定结果之间的接近程度。
溶液配制与实验要求同重复性。
6.4重现性
目的:考察在不同实验室,由不同分析人员测定结果之间的精密度。
溶液配制与实验要求同重复性。
7.准确度
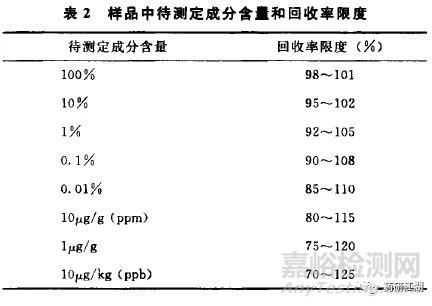
准确度系指分析方法的测定结果与真实值之间的接近程度,一般用回收率(%)表示。有关物质的准确度试验一般采用加标法,即:向原料药或制剂的供试品中加入已知量的杂质对照品进行测定。用至少测定6份结果进行评价,或设计3种不同浓度,每个浓度平行配制3份。计算时需扣除本底供试品的杂质含量。
2015年版中国药典关于回收率的可接受范围截图如下:
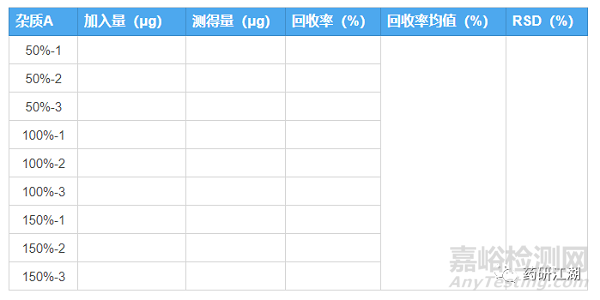
推荐结果整理表格如下:
注意事项:
a.在研究过程中杂质限度可能会有所变化(收紧或放宽),回收率的浓度设计可多个不同浓度,以便涵盖变动后杂质测定浓度范围。
b.试验中每一个供试品溶液的回收率均应满足回收率限度,而不是只看回收率均值。
c.有时回收率不合格,并不是因为无法回收,而是因为杂质峰型不好导致数据结果不佳,此时应合理积分或对色谱条件进行微调,尝试改善峰型。
8.耐用性
目的:考察色谱条件发生微小变化时对有关物质结果的影响。
按照拟定标准配制空白溶剂、对照品溶液/自身对照溶液和供试品溶液,计算不同条件下供试品的有关物质测定结果,并关注系统适用性是否符合标准要求。
同样放一张表格,将耐用性试验内容和结果要求直观呈现如下:
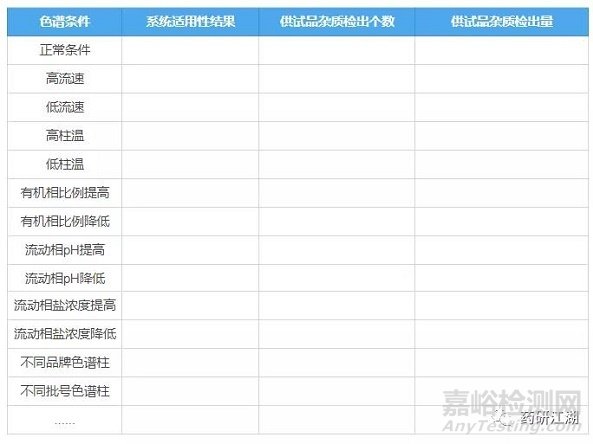
若耐用性试验中发现分析方法对某个条件要求苛刻,应在质量标准中予以体现,并注明可接受变动的范围。
9.其他
这个模块简单来说,就是质量标准规定什么,就去验证什么。
举例来说:规定了杂质的相对保留时间,就去探讨和验证相对保留时间的准确性和耐用性;规定了几分钟内的色谱峰作为空白辅料峰扣除,就去验证所扣除的时间段是否严谨准确。
三、总结和思考
每个有关物质的分析方法都有各自的特点,应对具体情况作具体分析。方法学验证是为了形成更科学和全面的分析方法,作为研究者,不能千篇一律,为了验证而做验证。遵循和严守是质量人员的职责,而开发与完善是研发人员的职责。在试验过程中发现问题、解决问题,并在每一次周旋与教训之后获得新的思考,这就是研发者在职业生涯中最珍贵的,经验。

来源:铭研医药


