您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-11-17 16:26
近期,国家药监局发布了《医疗器械临床评价技术指导原则》(以下简称《临床评价指导原则》)等5项技术指导原则,以加强医疗器械产品注册的监督和指导工作。众所周知,临床评价是基于医疗器械的适用范围,对产品安全性和有效性进行评价的一项重要活动。那么,你了解新版临床评价指导原则都有哪些变化吗?
Part 1 What
此次新版《临床评价指导原则》首先厘清了临床试验、临床数据、临床评价、临床证据几个关键词的基本概念,以及它们之间的关系。“临床试验”(Clinical Investigation)是为评价医疗器械的安全性、临床性能和/或有效性,在一例或多例受试者中开展的系统性的试验或研究。临床试验是获取“临床数据”(Clinical Data)的一种方式。“临床数据”(Clinical Data)是医疗器械临床使用过程中产生的安全性、临床性能和/或有效性信息。临床数据可以来源于多个方面,可以是申报产品或同品种医疗器械上市前和上市后的临床试验或文献数据,也可以是登记研究、不良事件数据库、病历数据等临床经验数据。“临床评价”(Clinical Evaluation)是由注册申请人采用科学合理的方法,对临床数据进行的分析评价,用于论证产品对安全和性能基本原则的符合性。临床评价后形成的报告及相关数据作为“临床证据”(Clinical Evidence)纳入技术文档中。“临床证据”(Clinical Evidence)是医疗器械技术文档的重要组成部分,注册申请人可根据现行法规的要求,提交临床证据以供监管部门审评。它们之间的关系如下图所示:
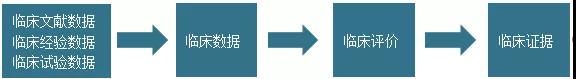
关于临床评价相关的几个概念,现在你清楚了吗?
Part 2 Why
一直以来,临床评价资料都是国内外医疗器械上市前审评过程中备受关注的重点。为什么各国监管部门都会如此关注临床评价呢?主要有两方面原因:一是就产品本身而言,医疗器械产品是以临床应用为目的,所以要关注临床的安全性及有效性;二是从监管角度出发,为了保证医疗器械的安全、有效,保障人民健康和生命安全。此次新版《临床评价指导原则》专门开辟篇幅详述临床评价的重要性,强调产品注册时需证明:
(1)在适用范围下,产品已达到预期性能;
(2)与受益相比,已知以及可预见的风险已降至最低并可接受;
(3)对医疗器械安全性、临床性能和/或有效性的任何宣称均可得到适当证据的支持。
临床评价是对产品设计开发的确认,是为证明所设计的产品安全并且满足预期用途而开展的活动,所以临床评价资料无疑是其中比较重要的证据。
Part 3 When
医疗器械“全生命周期临床评价”是此次新版《临床评价指导原则》反复强调的一个概念。临床评价是一个持续进行的活动,注册申请人需在产品全生命周期中进行周期性评价并更新临床证据。临床评价不再只是上市前的一个注册要求,注册申请人还需重视产品上市后的临床数据收集。
产品获批上市后,随着产品安全性、临床性能和/或有效性信息的不断更新,注册申请人还需开展器械上市后的跟踪、监督和持续评价,以及时跟进产品风险受益的重大变化。如需对禁忌证、警告、预防措施或说明书等方面进行变更时,需向监管机构申请变更注册、说明书更改告知等。
Part 4 Who
临床评价如此重要,谁才有资格担任临床评价人员呢?新版《临床评价指导原则》明确规定,临床评价人员需具有专业水平和经验,包括熟悉产品技术及其使用;掌握临床研究方法,如临床试验设计、生物统计学等;了解预期诊疗疾病的诊断和管理知识等各方面能力。
此外,临床评价报告中需要有专门章节论述临床评价人员选择的合理性。
敲黑板,临床评价工作可不是随便就能开展的哦!
Part 5 How
本次新修订的《医疗器械监督管理条例》引入了灵活多样的临床评价模式,包括免于进行临床评价的情形。
而新版《临床评价指导原则》则进一步对条例中的要求,从实操层面进行了明确和细化。
下面就跟着小U看看不同的临床评价路径,在注册阶段需要对应提交哪些资料吧!
路径一:免于进行临床评价
适用产品类型:列入《免于进行临床评价目录》的产品
所需提交资料:
在“非临床研究资料”部分提交以下文件:
1)申报产品相关信息与《目录》所述内容的对比资料;
2)申报产品与《目录》中已获准境内注册医疗器械的对比说明和相应支持性资料。
路径二:通过同品种医疗器械临床数据进行等同性论证
适用产品类型1:通过等同器械的临床数据进行临床评价
所需提交资料:
医疗器械临床评价报告,重点阐述以下内容:
1)申报产品与对比器械等同性论证;
2)若存在差异,需证明具有相同的安全有效性;
3)等同器械临床数据的总结与评估;
4)等同器械临床数据的分析等。
适用产品类型2:通过可比器械的临床数据支持申报产品的部分临床评价
所需提交资料:
医疗器械临床评价报告,重点阐述以下内容:
1)申报产品与对比器械可比性论证;
2)可比器械临床数据的总结与评估;
3)可比器械临床数据的分析。
注:若差异性不能通过申报产品的非临床研究资料、临床文献数据、临床经验数据证明对产品安全有效性不会产生不利影响,则需开展针对差异性临床试验。
路径三:临床试验
适用产品类型:已有临床文献资料、临床数据不足以确认产品安全有效,必须通过临床试验进行临床评价的产品
所需提交资料:
医疗器械临床评价报告(格式要求见《医疗器械注册申报临床评价报告技术指导原则》附件)
附件至少包括:
1)临床试验方案;
2)各临床试验机构的伦理批件;
3)经批准的知情同意书/患者须知文件样稿;
4)病例报告表样稿;
5)临床试验报告;
6)临床试验数据库(原始数据库、分析数据库、说明性文件和程序代码)。
备注:
同品种医疗器械,包括:等同器械和可比器械两种情形。
等同器械:相同的适用范围、相同或相似的技术特征和生物学特征。
可比器械:相似的适用范围、相同或相似的技术特征和生物学特征。
此次更新完善的《临床评价指导原则》,将医疗器械全生命周期临床评价纳入其中,对临床评价人员提出了更高的要求,评价模式更加科学,更加多样化,有利于进一步节约企业、医疗机构和监管部门等主体的资源,提高效率,从而进一步鼓励创新。作为注册申请人,我们需要认真研读法规,基于产品的实际情况,灵活审慎地选择合适的临床评价路径,踏实做好全生命周期的临床评价。

来源:质量法规那些事


