您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-09-17 05:56
1.前言
蛋白多肽、抗体以及核酸类药物等都属于生物技术药物(Biotech drugs),近年来蛋白多肽类药物应用愈加广泛[1]。蛋白多肽类药物与小分子药物相比,优势在于蛋白多肽药物特异性更强,副作用更小,因为无论是经过生物工程设计还是化学合成的,它们的组成和代谢通常都与内源蛋白质相同或相似[2]。而且,此类药物与靶标结合的特异性更强,这就意味着与非靶标结合率极低甚至不结合[3]。目前小分子药物研发已经到了瓶颈期、某些特定疾病小分子药物已不能达到治疗目的,挖掘生物技术药物的潜力变得尤为关键。蛋白多肽类药物膜通透性差且对酶极为敏感,所以这类药物一直以来在临床上都以注射给药为主,但是注射剂生产成本高,注射途径使用不便、费用高、长期用药增加患者的精神负担,极大的降低患者依从性,有些药物也会出现副作用[4]。然而,有报道称,此类药物的副作用通常是由于注射部位的剂量较大造成的,通常为局部不良反应。例如,采用贝伐珠单抗(Bevacizumab)治疗年龄相关性黄斑病变需要频繁进行玻璃体内注射,易于诱发白内障、视网膜出血和脱离等并发症[5]。基于此,药学工作者一直致力于寻找患者更易于接受、毒副作用更小的新型蛋白多肽类药物的非注射给药途径,包括肺部、鼻腔、口腔、眼内、透皮、直肠和口服给药等。
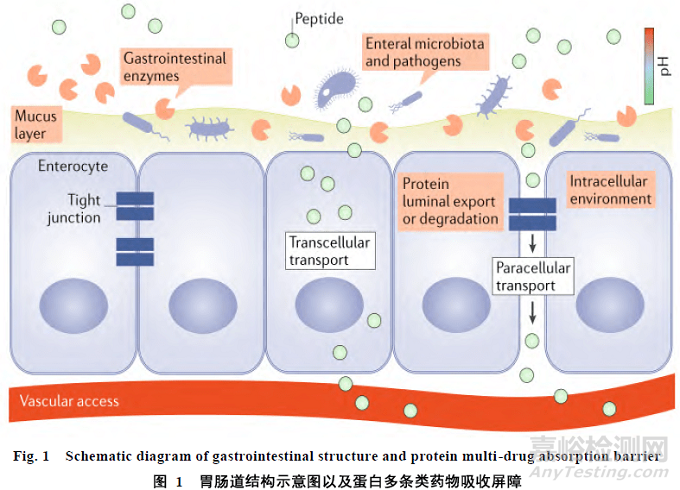
每种非侵入性给药方式都有其独特优点,但是多种给药途径相比较而言,口服给药最方便,特别适合于长期用药的患者,口服蛋白多肽一直是制剂开发的热点研究方向。蛋白多肽类药物的口服吸收需要克服强酸环境、水解酶、肠黏膜屏障[6-7]、首过效应等诸多不利因素[8],如图 1 所示[9],目前研究大都着重解决蛋白质体内稳定性和吸收困难的问题。Agyei 等[10]较详细的剖析了制剂开发的驱动力与限制因素,详见表 1。

从表中可以看出,开发蛋白多肽类药物的口服剂型存在重重困难,但科研人员长期以来的不懈努力获得了几种解决办法[11-12],包括化学修饰、添加吸收促进剂以及应用微粒给药系统等。随着美国食品和药物管理局(Food and Drug Administration, FDA)对蛋白制剂批准率的提高,用于治疗人类疾病的蛋白质产品的开发速度稳步增长,目前已有多种生物技术药物上市[13],如:口服干扰素、艾塞那肽、胰岛素等。蛋白制剂的开发,科研工作者关注生物利用度、靶向性、体内稳定性以及毒副作用等的同时,因其半衰期短,也需注意长效制剂的应用,防止药物出现波峰波谷现象,影响疗效。
2.蛋白多肽类药物制剂开发策略
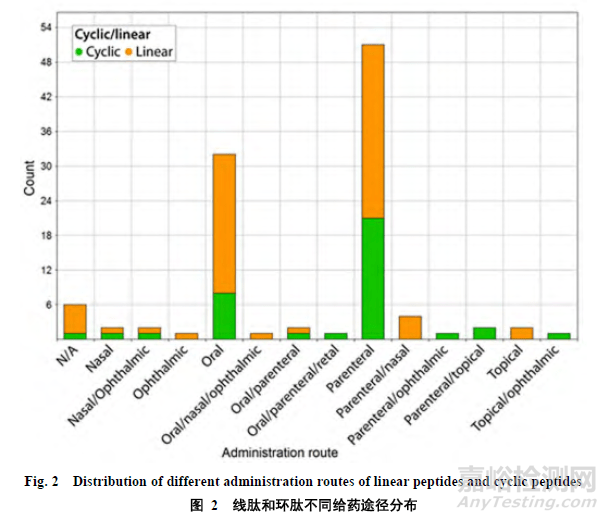
对于蛋白多肽类药物的开发,有学者指出:在药物设计上可能不像其他小分子药物一样遵循里宾斯基无规则[14],并不像人们想象的那样线状多肽一定优于环状多肽。从结构的角度来看,多样性是肽类药物的一个特性,因此,这些化合物的大小和复杂程度不应影响它们获得临床试验和进入市场的机会。同时,有一项统计显示[15]:从 2012 年到 2016 年,美国 FDA 批准的多肽类药物中,口服给药途径占有不小比例,见图 2。基于此,我们主要研究蛋白多肽类药物在口服给药途径上的改进和应用。
2.1 蛋白结构保护
为了提高蛋白质的体内稳定性,研究者们对蛋白质结构进行修饰改造,常见的方法有[16]:氨基酸替换、环化、PEG 化等。例如,牛源无基质血红蛋白(Hb)是用于载氧血液代用品的理想材料,PEG 修饰使得 Hb 的分子量增大,既有效延长在血管内滞留时间,又可降低肾脏消除速率[17]。化学操作对小分子药物很有效,但是对于分子量较大的蛋白多肽来说,特定结构对生物活性很重要,修饰往往不能达到预期的效果[18]。蛋白酶抑制剂既可以保证蛋白多肽原有结构,又可以使蛋白免遭酶破坏。蛋白酶抑制剂的应用取决于靶点的位置和细胞分布,如抑肽酶,是胰蛋白酶的一种,主要作用是可逆性抑制体内胰蛋白酶,可用于增强口服胰岛素微球的生物利用度[19]。然而,使用蛋白酶抑制剂可能会导致细胞酶活性丧失或过度激活,导致严重的副作用,如食物消化不良和胰腺肥大或增生[20],因此,在选择蛋白酶抑制剂时需要谨慎。
2.2 添加口服药物吸收促进剂
吸收促进剂是能可逆地、特异性或非特异性地显著增强胃肠道吸收,进而起到提高血药浓度和生物利用度作用的一些制剂材料[24]。这些物质可以不同程度地与上皮细胞膜脂或蛋白质相互作用,开放或破坏紧密连接的完整性,降低粘液粘度,或增加膜流动性。优良的吸收促进剂应取决于以下条件是否合适:作用的时间、渗透增强的程度、无毒性。几类公认的化合物(螯合剂、表面活性剂、胆盐、脂肪酸及其盐类)是优良的吸收促进剂,每类化合物都有其作用机制,如表 2[25]。

传统的促进剂主要是表面活性剂类,许多化合物具有潜在的吸收促进作用,但是毒性较大,导致胃肠道粘膜显著的、不可逆的损害[26]。近年来,卡波姆、壳聚糖及其衍生物成为研究的新热点。壳聚糖、卡波姆等本身无毒性,其生物黏附性及促吸收作用广泛应用于口服蛋白多肽类药物吸收的研究,其中壳聚糖及其衍生物的效果优于卡波姆。卡波姆因为携带大量羧基可在肠道内释放质子,一定程度上抑制蛋白酶活性;也有学者指出,卡波姆可以络合钙离子,提高细胞通透性,促进蛋白多肽类药物的吸收[27]。壳聚糖具有打开小肠上皮细胞间紧密连接的功能,使大分子药物更易越过上皮组织、增加药物在小肠内的吸收,同时肠内的其他物质也会趁着打开的通道进入体内。如果长期使用,不得不考虑毒副作用。据此有研究表明,羧化壳聚糖可以降低毒性,尹丽芳等[28]制备了牛血清白蛋白-羧化壳聚糖纳米粒,通过体外实验发现:相比于牛血清白蛋白-壳聚糖纳米粒,使用羧化壳聚糖的纳米粒,吸收更好,毒性更低。
2.3 微粒给药系统
微粒给药系统包括微球、微乳、纳米粒等,在此并不特指某一种,粒径为 1~250 μm 的粒子称为微球,粒径在 0.1~1 μm 的粒子称为亚微粒,10~100 nm 的粒子称为纳米粒[29]。将蛋白多肽类药物装载于微粒载体上,一方面可以保护蛋白,防止胃肠道的强酸性环境和消化酶对药物的破坏,另一方面可以利用载体材料达到长效释药或靶向治疗的目的[30-31]。通常,微米级别的粒子易被淋巴结摄取。因此口服微球可以靶向到胃肠道的淋巴结,继而跨膜转运吸收进入体循环,而且利用特殊载体材料能制备生物黏附功能性微球,有效提高蛋白多肽类药物的生物利用度。
在微粒给药系统中,微粒的材料、表面特性、粒径等多项因素对微粒存在影响[32-33]。所选材料应该可以包裹药物,保护其免受消化系统中酶的降解;纳米粒的大小、形状及其分布要符合要求;有较高的载药量及包封率;药物的释放时间应较好地足临床用药标准;载体材料必须是无毒、可降解的。常用的纳米载体材料可以分为两大类[34]:①合成高分子材料,例如聚丙交脂、聚丙交酯乙交酯共聚物、聚己内脂等。②天然高分子材料,包括明胶、海藻酸盐、壳聚糖、蛋白类。材料本身对纳米粒径及释药速度有很大影响。天然高分子材料的降解比较快,释药速度也快。聚酯单独或组合在蛋白质输送方面的应用研究最为广泛,与天然聚合物相反,合成聚合物可以实现在几天到几周的时间内调控药物释放。
2.4 靶向给药
人们已经研究了许多方法来克服口服疗法的局限性。在这些方法中,靶向给药系统的设计和开发是为了克服新发现的药物和传统剂型的缺点。通过口服途径给药, 靶向给药系统(DDSS)通过提高药物分子的口服生物利用度和降低药物的最低有效浓度来减少与剂量相关的毒性,提高治疗效果。
被动靶向是通过利用细胞的温度、pH、异常血管或表面电荷等病理生理学特征来增加目标部位的积聚来实现的[35]。然而,靶向的随机性及其不足或非特异性严重限制了其应用。而在主动靶向优势在于,特定的配体连接在药物或者载体的表面,将整个系统引导到表达配体特异性生物标志物的特定组织/位点。抗体、维生素、叶酸以及多肽等都可以作为靶向给药的配体[36]。配体与存在于靶部位表面的受体相互作用,从而导致递送系统在靶部位积累,药物既可以表面作用,也可以内化[37]。其中维生素 B12 和叶酸(VB9)是研究最多的配体,维生素 B12 为水溶性维生素,口服后先与胃中的一种唾液酶(Haptocorrin)结合成复合物,在 Haptocorrin 保护下,维生素 B12 进入小肠,维生素 B12 再与肠上皮细胞表面的内在因子结合进入体循环[38]。
各种吸收不良的药物、蛋白质和多肽(如胰岛素),以及其他分子,通过使用维生素 B12 作为靶向递送的配体,大大提高了它们的口服生物利用度。维生素 B12 修饰的含有 4%(w/w)胰岛素的葡聚糖纳米粒,与非靶向纳米粒相比,其生物利用度显著提高(2.6 倍)[39]。值得注意的是,与其他维生素相比,维生素 B12 的吸收相对较慢,吸收部位有限(主要在回肠末端),因此维生素 B12 的应用受到了影响[40]。叶酸与维生素 B12 相比,受体数量足够多,并通过改善生物活性分子或囊泡系统的摄取和运输来加强吸收。据报道,各种吸收不良的药物,如蛋白质药物(胰岛素)、抗生素(万古霉素)和抗癌药物(多西紫杉醇和紫杉醇),通过特异性叶酸受体靶向已显示出显著的生物利用度[41]。
2.5 定位给药系统
组成胃肠道上皮的细胞种类繁多,呈现不同的特性和受体,尤其是在小肠。其中有研究可以作为药物吸收部位的细胞主要有:肠上皮细胞(Enterocyte)、杯状细胞(Goblet cell)以及 M 细胞(M cell)等,如图 3 所示。以胃肠道上皮为靶点的 DDSS 是口服给药中最常用的给药策略之一。这一策略在很大程度上被用来提高难溶性药物或生物制品的生物利用度,或增加药物递送系统在靶部位的累积[42]。
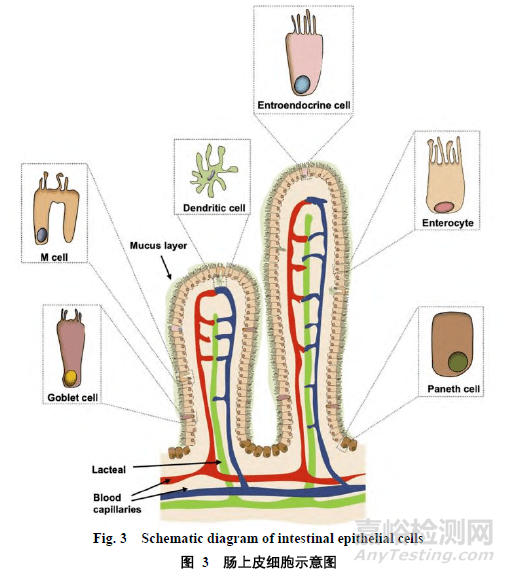
2.5.1 杯状细胞
杯状细胞约占小肠细胞总数的 16%,负责粘蛋白的产生、储存和持续释放,粘蛋白是粘液层的主要组成部分。它们具有保护性屏障功能,因此极大地限制了治疗药物的吸收。尽管这种细胞系很丰富,但杯状细胞很少被用作靶点,到目前为止,多肽 CSKSSDYQC(CSK)是唯一被证明可以作为口服给药的配体[43]。
M 细胞是一种特殊的上皮细胞,是粘膜免疫系统的一部分,是微生物入侵的入口,参与特异性免疫和食物耐受性[44]。这些细胞主要存在于肠道相关淋巴组织的滤泡上皮,它们有运输颗粒物的能力,包括抗原、细菌和病毒[45]。此类细胞区域水解酶活性低,分泌粘液的杯状细胞有限[46],从而使得 M 细胞有效地摄取蛋白多肽药物。虽然 M 细胞占肠道细胞不到 1% 的数量,由于其上述特性,这些细胞是口服药物递送极有希望的靶点。研究表明,特异性 M 细胞靶向可以弥补口服给药中 M 细胞数量少的局限性[47]。有研究者制备了 CKS9(一种 M 细胞归巢九肽)-水溶性壳聚糖-PLGA 微球,用于包裹猪痢疾杆菌的膜蛋白,口服后,微粒能够提高系统的 IgG 抗体应答[48]。
ATB 0,+ 是位于结肠的管腔表面的一种氨基酸转运蛋白,已被作为口服给药的靶点进行了研究。在 Na + /Cl − 梯度的驱动下,ATB 0,+ 可以运输大多数中性和阳离子氨基酸[49]。但目前对于多肽药物在此处的吸收研究较少。虽然结肠的表面积不适合吸收,但环境中几乎没有消化酶,药物停留时间长。因此,结肠可能对多肽的吸收有一定的作用。时间控制型、包衣剂型或者其他类型的结肠定位给药系统,可以将制剂靶向输送到结肠[1]。通过对肝素的研究,Muranishi 提出[50],当混合胶束用于促进吸收时,结肠比小肠更有利。
2.6 其他
为了维持蛋白多肽药物在体内的活性和(或)保证生物利用度,除了使用以上方法,还可以考虑使用细胞穿透肽、新型辅料的应用、肠溶包衣等手段。
3.口服蛋白制剂简介
3.1 口服长效片剂
为延长蛋白药物的体内半衰期, 研究人员通常采用长效化手段[51]、PEG 修饰、缀合脂肪酸链、环化或氨基酸替代等策略,并已有多款药物成功上市。除对蛋白质及多肽进行分子改造外,还可通过制剂学手段改善蛋白及多肽类药物的吸收并使其长效化。在人体中,药物通过小肠的时间可能非常快(从胃到结肠平均需要 3 小时),在十二指肠的停留时间可以限制在几分钟[52]。因此,蛋白多肽的吸收不良也可能归因于吸收表面和药物之间的接触时间较短。
口服制剂实现长效缓释作用,需要延长其在消化道中的停留时间并避免药物被降解。VivekGupta 等[53]受透皮贴剂的启发,设计了一种黏膜黏附片,其作用机制见图 4。

首先制备含药的肠黏附片,并在其一侧用水不溶性材料进行包衣,待其到达肠道内,未包衣处的黏附层通过氢键等作用力与肠黏膜结合,而水不溶的包衣层对肠道中多种蛋白酶有物理隔离作用,可防止药物降解。例如,采用卡波姆、羧甲基纤维素钠物理混合物为肠黏附片基质,在单侧包裹乙基纤维素背衬层,可使鲑鱼降钙素的口服吸收显著提高[54]。类似的结构还被用来口服递送胰岛素和艾塞那肽[55],均能有效增加药物的口服生物利用度。
3.2 口服微粒
口服小粒径、正电荷且具有粗糙表面的微粒更易于在胃肠道滞留和被肠道吸收[56];而肠溶包衣颗粒则可有效避免胃酸破坏。基于此,研究人员采用聚乙烯亚胺混合胰岛素制备纳米粒,并依次涂覆水溶性聚合物羟丙甲纤维素、混有崩解剂羧甲基淀粉钠的聚甲基丙烯酸酯(Eudragit ® NE)和 Aqoat ® ,达作用时间 48 h 以上;此外,Zhao 等[57]采用 SiO2 作为胰岛素的口服缓释载体并在外包被肠溶材料,能够在 27 h 内有效控制血糖水平。有人[58]采用一步喷雾干燥技术制备 β-环糊精微粒,并通过在处方中加入肠溶性阻滞聚合物增加制品的稳定性,用其作为口服胰岛素载体。载药微球的平均粒径为(0.08~0.25)μm,体外释放实验表明,胰岛素在人工胃液中不释放,在人工肠液中 8 h 可释放 50%,可见该微球可以免受胃液破坏,在肠液中释放较好。另外也有研究表明,生物利用度还与粒径分散度有关,Panayiotis P[59 ]等制备了 RGD 多肽微乳(平均粒径0.1~1 μm,分散度 0.2~0.6),其结果表明,生物利用度的提高与微乳成分有很强的相关性,而与粒径并没有明显的关系。
胃肠道上皮细胞的绒毛结构因表面积较大,为一些高分子材料黏附提供了足够的着力点,因此可利用生物黏附性延长药物在靶部位的停留时间[60],而且制剂与黏膜绒毛紧密接触能增加药物的跨膜吸收速率[61]。常见的生物黏附性材料有卡波姆[62]、羟丙基甲基纤维素、甲基纤维素等。壳聚糖及其衍生物兼备黏膜黏附性和促透作用。利用生物黏附性材料制备口服微球,不失为一个提高蛋白药物生物利用度的好办法。在一项研究中,羧化壳聚糖接枝甲基丙烯酸甲酯纳米粒已被开发用于口服胰岛素[63]。该纳米载体在较低的 pH(2.0)下相对于较高的 pH(6.8 和 7.4)表现出缓释作用,具有良好的组织相容性和血液相容性。
聚合物微球已用于口服疫苗制剂。例如,霍乱弧菌全细胞口服 PLGA 微球疫苗,该微球含有来自霍乱弧菌的全细胞裂解物[64]。流行性乙型脑炎病毒疫苗也是通过将其装载于可生物降解的PLGA 微球中来制备的[65]。
3.3 口腔给药
为克服生物利用度低的问题,一种试图完全避免胃肠道的给药策略,研究者们试图将药物从口腔直接运送到血液中。更多地了解口腔的解剖学和生理学有助于设计合理的给药策略,口腔由几个部分组成,主要包括唇、硬腭和软腭、牙龈、舌以及舌下部位。药物吸收途径为从口腔内静脉进入颈静脉再进入体循环[66]。颊粘膜和舌下粘膜常作为给药部位,因为它的渗透性比其他区域要高。颊粘膜和舌下粘膜具有非角化表面、非极性脂质和细胞间脂质的无定形组织,这种组织对多肽类药物通过细胞旁途径扩散的阻力较小。舌下途径主要用于输送需快速起效的小分子药物,如硝酸甘油。相比之下,口腔途径具有较小的表面积和较低的渗透性,更适合于作用时间较长的蛋白多肽类药物。通过口腔途径给药的主要挑战是唾液及其相关代谢酶的活性,几种药物,包括胰岛素、胰岛素原、脑啡肽类似物、促甲状腺激素释放激素和降钙素,在与动物口腔组织匀浆孵育时都会降解[67]。为了阻止酶降解,口腔途径给药可以向制剂中加入酶抑制剂,如胆盐、抑肽酶等。生物粘附片、贴片、软膏和粉末,已被用于蛋白多肽的口腔给药[68]。在此,值得一提的是口腔膜同样作为一种创新的、非侵入性的方式,以最大限度地顺应患者,特别是老年人、儿科或有吞咽并发症的患者,提供生物活性蛋白或肽。长效口腔膜[69]可以减少给药次数,以免患者因忘记时间而影响药物疗效。
4.总结与讨论
经过几十年的发展,目前此类制剂研究仍主要集中于植入剂和注射用微球的开发。为提高蛋白质稳定性和生物利用度,科研人员采用不同技术手段,除了化学修饰以外,应该更多的思考制剂学手段,以期为患者提供更多的治疗方案。综合考虑研发、生产成本、使用成本和患者依从性等众多问题,口服蛋白多肽类药物有一定的优势。但是又考虑到蛋白多肽适宜水环境,被制成固体制剂,也会面临不小的难度。口服蛋白多肽类药物新剂型的开发离不开新型药物载体材料的研发,近年来越来越多的生物材料得以广泛应用,这也为蛋白多肽类药物开发提供了可能。总之,蛋白及多肽类药物的递送具有重要临床意义和广阔的市场前景。
当然,目前受众多限制因素影响,口服蛋白多肽类药物尚存在一些潜在问题。例如,目前大部分研究主要集中于如何提高蛋白多肽类药物体内稳定性,对于此类药物在体内的释放行为的研究相对较少;吸收促进剂可以增加药物的吸收,但是对蛋白质没有特异性,在促进药物吸收的同时,也可能促进胃肠道毒素和消化酶进入血液;黏膜黏附系统可以延长蛋白多肽药物在胃肠道的滞留时间,但不能保证药物的通透性,也不利于小肠粘膜的清洁;口腔给药虽然能避免胃肠道,但是能成功应用的药物数量有限;对于患有胃溃疡、胃肠炎的患者因胃肠道环境改变,此类药物的效果值得考虑;实现大规模工业化生产的品种有限。
展望未来,伴随着科研力量投入的不断增多,相关研究理论日臻成熟,蛋白类药物的开发技术也不断进步,这不仅能够改善药品的治疗效果,也能优化生产过程,提高蛋白类药品的产量与质量,解决产能不足导致价格居高不下和因技术不过关使得药物质量差的问题,为临床上普及应用创造良好的条件。

来源:搜狐网


