原料药作为一种特殊商品,其质量管理具有特殊性,如何平衡执行力与持续改进,如何在满足标准要求的前提下实现卓越,对企业的管理者来说是一项新的课题。本文结合实际工作,整理总结原料药洁净区环境管控的程度,以期帮助同行更好的理解相关法规。
原料药作为制剂中的有效成分,对制剂的安全性和有效性起着决定性的作用,因此不管是在我国,还是在欧美发达国家,甚至于在原料药制造大国印度,原料药的生产质量管理要求一直都是参照制剂的要求进行管控。同样,洁净区的管控也是照搬制剂企业的管控要求。在原料药企业飞速发展的近几年,由于各类检查人员的认知不统一,检查的尺度不统一,作为原料药生产企业的质量管理人员,有时会受困于如何把握这个度的问题。
从特性上来看,原料药分为无菌原料药和非无菌原料药,无菌原料药需要比较严格的生产环境条件,这一点在行业内应该是能达到共识的,也不会有企业老板或者负责人敢对无菌原料药的生产环境降低要求。那么对于非无菌原料药呢?标准太严,企业运营成本就高,标准太松,又会被客户挑战,被监管部门挑战。所以太严或者太松,对于企业来说都不能接受,那么到底应该如何管控?该参考哪些标准呢?
根据中国的《药品生产质量管理规范》(即GMP)的规定,药品生产需要在时宜的环境中进行,并且GMP原料药附录部分对于原料药生产环境也给出了指导原则,规定非无菌原料药精制、干燥、粉碎、包装等生产操作的暴露环境应当按照D级洁净区的要求设置。也就是说需要控制生产环境的温湿度、压差以及悬浮粒子等。
ICHQ7规定,应当根据需要,配制足够的通风、空气过滤和排气系统,应视生产步骤的要求,设置包括控制气压、微生物、灰尘、湿度和温度的设备。这里规定的就比较灵活,让企业根据风险分析,自行评估需要配制什么样的环境控制设备,以及生产环境需要控制到什么水准。只要你最终出来的原料药产品符合安全、有效、质量可控即可。
我们知道,产品的储存温湿度可以根据稳定性实验数据获得,那么生产环境的温湿度如何控制呢?为了控制成本,原料药生产企业一般控制温湿度也只是控制产品最终成型的步骤(即内包装前一步),要求产品暴露的环境尽量和产品的储存温湿度相一致,比如产品贮存温度要求是阴凉,该区域的环境就按照阴凉的标准设置,同时也要考虑操作人员的舒适性,比如在冬季,环境温度较低时,甚至达到零下,如果不采取温控措施的话,操作员工必定会感到不舒适,就不能保证产品持续稳定的生产。这里面还有一个问题,就是不同国家对温度的定义不同,企业可以根据产品销售市场的不同,选择合适的控制温度,以下列举了常见国家对常见温度的定义:
同样,对于湿度,既要考虑产品的特性,如吸湿性,也要考虑人员的舒适性。笔者所在的公司曾生产的某原料药因吸湿性较强,稍有不慎,产品水分就超标,产品检验不合格导致报废的情况时有发生,研发部门就要求将生产环境相对湿度控制在30%RH以下,果然产品合格率立马就提升了,但由于湿度较低,员工在操作过程中难免有抓挠身体的情况,再加上穿戴不符合要求,最终接连发生几起因外来物导致的客诉投诉。同样,环境湿度亦不可过大,否则产品可能产生霉变,据某研究机构研究结论,当药品暴露在环境湿度80%RH条件下一段时间后,不论环境温度高低,药品储存过程中后均会发霉。
对于压差,一般的要求是相同洁净等级的不同操作功能间压差在5Pa以上,不同洁净等级的功能间压差在10Pa以上。这个要求在监督部门和生产企业间应该都是能达到共识的,但根据经验,空调实际运行过程中,大部分企业都是在产品生产间歇期,关闭空调系统,待生产开始前再打开空调系统,这时就会发现某两个功能间之间的压差不符合要求,在排除过滤器破损和堵塞的情况下,无法确定总送风量是否需要增加时,可以尝试调整回风口的大小,从而达到增加压差的目的,但因调整某个区域的压差,可能对其他功能间之间的压差产生影响,一般按洁净度从高到低逐级调整。另外,对于洁净区内的门的开向,也遇到一个很有意思的事,在和某新建企业的品质人员交流的时候,对方提到他们遇到一个难题,就是洁净区压差调整到符合要求后,发现过几天又变差了,经过实地参观,发现他们的洁净区内房间的门设计的有问题,是从高压朝低压房间方向开,从而导致门经常会被压差挤压,关不严实,这样不仅仅是压差不足的问题了,还有可能造成交叉污染。
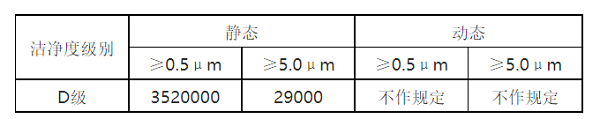
对于悬浮粒子,其为评判生产环境是否为洁净区,以及洁净区等级的关键指标,一般企业都会按照规定的频次进行测试,检查时也是必看项目。按照D级(相当于ISO 8级)洁净区要求进行控制,即如下表格,测试方法一般参照ISO14644-1标准。
合理的空间布局和良好的操作规范是确保原料药洁净区环境持续达标的根本保障,原料药生产洁净区环境控制是一门学问,做的好,员工舒适、老板省钱、检查官认可、客户满意,做不好,将会事倍功半,品质人员也会被“问责”。