引言
药品变更管理贯穿研发、临床、生产、流通、上市后研究等全生命周期,在创新药物的临床研究期间,药学变更是不可避免的重要环节。在进行药学变更时,临床试验的安全性和数据的可靠性是首要考虑因素。任何药学变更都需要经过科学严谨的评估和审查,以确保对患者的治疗效果和安全性没有负面影响。此外,在药学变更过程中,需要进行必要的数据记录和文档整理,确保变更过程的追踪和监控,并通过适当程序(补充申请、DSUR(研发阶段安全性更新报告))及时通报监管部门。总的来说,创新药物的临床期间,药学变更是为了不断优化和改进药物,提高临床疗效和安全性。但在进行药学变更时,必须遵循严格的规定和指导,以确保变更的安全性和数据的可靠性。
一、创新化药临床期间与上市后药学变更管理对比
通过对《创新药(化学药)临床试验期间药学变更技术指导原则(试行)》、《已上市化学药品药学变更研究技术指导原则(试行)》中有关内容的比较,可以看出,在这两个文件中,既有相似之处,又有不同之处。两者都是采取以安全风险评价为基础,由申请者进行风险评估并确定风险等级,但是其管理方法上却有所不同。
1.1 变更分类和要求
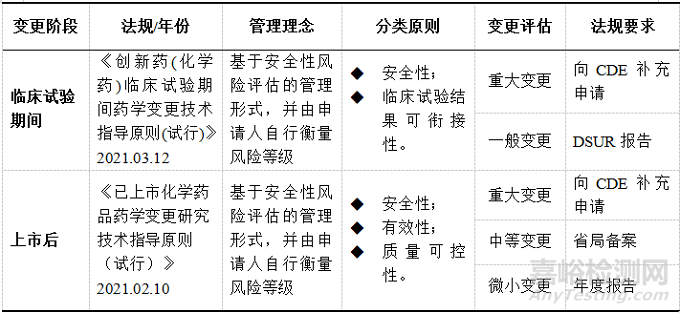
临床试验期间药学变更按照对临床受试者安全性、临床试验结果可衔接性影响风险分为重大变更、一般变更两类进行管理。上市后药学变更按照对产品安全性、有效性和质量可控性的影响分为重大变更、中等变更、微小变更三类进行管理,见表1。
▲表1-创新化药临床试验期间和上市后药学变更分类和要求
注:早期临床研究阶段,药物的人体安全性尚未完全确立,重点评估药学变更对于受试者安全性可能产生的影响;关键临床研究阶段,除需重点关注受试者安全性外,还需兼顾临床试验结果的可衔接性。
1.2 变更程序
申请人评估认为可能增加受试者安全风险的变更,应当按《药品注册管理办法》提出补充申请,认为不影响受试者安全的,可以直接实施并在研发期间安全性更新报告或在上市后年度报告中说明。
(1)临床期间变更
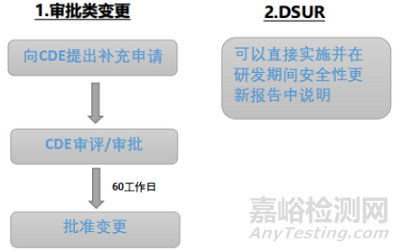
与上市后药品变更不同,对于临床试验期间的药学变更,CDE把评估权更多的交给了申请人。只有影响受试者安全性的重大变更要向CDE提交补充申请,如果临床期间的药学变更属于未影响受试者安全性的一般变更,申请人可以直接实施并在DSUR报告中说明,见图1。
▲图1-创新化药临床试验期间药学变更程序
(2)上市后变更
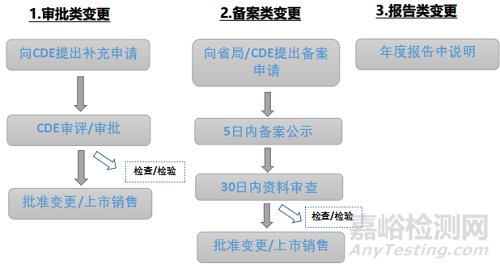
根据法规要求药品上市后变更程序主要包括审批类变更、备案类变更及报告类变更,见图2。
▲图2-创新化药上市后药学变更程序
其中CDE审批类变更申请审评时限为60工作日,补充申请合并申报事项的,审评时限为80工作日,其中涉及临床试验研究数据审查、药品注册核查检验的审评时限为200工作日。
1.3 补充申请资料
根据2021年2月10日国家药监局发布《已上市化学药品变更事项及申报资料要求》附件,国家药品监管部门审批的补充申请事项包括:
国家药品监管部门发布的已上市化学药品药学变更相关技术指导原则中属于重大变更的事项。
国家药品监管部门发布的已上市化学药品临床变更相关技术指导原则中属于重大变更的事项。
药品上市许可持有人主体变更。
使用药品商品名。
国家药品监管部门规定需要审批的其他事项。
从上述规定来看创新药临床期间和上市后药学重大变更均纳入国家药品监管部门审批补充申请管理事项。
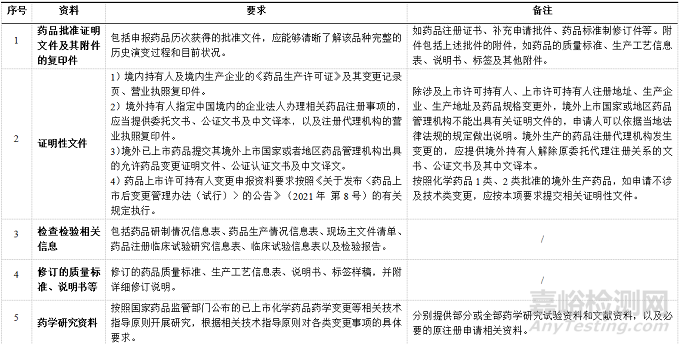
药品上市许可持有人应根据所申请事项,按表2所示,整理并提交申报资料,不适用的项目应注明不适用并说明理由。
▲表2-创新化药补充申请资料
二、临床试验期间药学变更分类与研究内容
创新化药临床期间药学变更包括原料药和制剂两大类。对于原料药变更,需要考虑生产场地、工艺、质量标准、包装容器和贮藏条件等。而在制剂变更方面,则需要注意生产场地、处方工艺、辅料工艺、剂型与规格、质量标准、包装容器和贮藏条件等。申请人通过详细的研究和测试,确保变更后的药品仍满足临床试验的要求,并保证受试者的安全。
2.1 原料药变更
对于原料药变更研究的一般原则,是需结合其对相应制剂的质量影响开展评估和研究。针对自身产品特点,根据风险评估结果开展研究。如出现变更前后产品质量不可比的情况,申请人需要通过前期的数据积累进行风险评估,合理的完成桥接研究。
▲表3- 临床试验期间原料药药学变更分类与研究
对于复杂分子(如:合成多肽、小分子核酸等)、复杂工艺(如:发酵类、生物来源提取物类等)原料药的变更,其对临床样品的潜在影响的体外评估手段相比化药更具特异性,需在遵照本指导原则研究思路的基础上,结合产品自身特点开展充分的风险评估及变更研究工作,以获得更多的变更支持性数据。
2.2 制剂变更
对于制剂变更,需重点从变更对于药物的制剂性能、安全性相关指标的影响来展开评估和研究。
▲表4- 临床试验期间制剂药学变更分类与研究
一般而言,变更制剂剂型、规格、处方、生产工艺、包装和贮藏条件等都有可能会对制剂性能产生影响,在变更研究中需结合药物性质、剂型特性、处方工艺特点以及变更的具体情况评估变更对制剂性能可能产生的影响,根据风险评估结果,选择适宜的制剂性能相关指标开展变更支持性研究,必要时还需考虑进行制剂体内桥接研究。
对于安全性相关指标,杂质种类和杂质水平是影响临床试验药品安全性的重要因素。对于注射剂、吸入制剂、植入剂等而言,还需关注无菌、细菌内毒素(或热原)、不溶性微粒(真溶液型注射剂)、可见异物(真溶液型注射剂)、渗透压摩尔浓度(注射剂)等。需确保变更后产品的相关指标仍符合安全性要求。
三、结束语
申请人在创新化药临床期间进行药学变更研究时,申请人需要明确变更的原因、事项和程度。同时,要结合品种特点和具体的变更内容,按照风险评估的思路来评估变更可能对药品质量、临床试验受试者安全性和临床试验结果可衔接性产生的影响。在评估的基础上,可以判断变更是重大变更还是一般变更,并相应地展开研究工作。此外,药学变更往往不是孤立发生的。例如,在进行生产场地变更时,可能伴随着生产设备和生产工艺的变更;在进行处方变更时,可能引发药品质量标准的变更或与之相关。对于多个变更同时发生且存在关联的情况,可以根据《创新药(化学药)临床试验期间药学变更技术指导原则(试行)》的基本思路,分别展开研究工作。重点关注技术要求较高的变更类别,并注意多个变更可能带来的叠加影响。
总之,进行药学变更研究时,申请人需全面评估变更的影响,确定变更的风险等级,合法、合规地展开相应的研究工作,确保变更后生产的药品符合质量要求、保证临床试验受试者的安全,并能够产生可靠的临床试验结果。
参考文献
[1] 《创新药(化学药)临床试验期间药学变更技术指导原则(试行)》(2021年第22号).CDE,2021-03-12.
[2] 《已上市化学药品药学变更研究技术指导原则(试行)》 (2021年第16号).CDE,2021-02-10.
[3] 《药品注册管理办法》(2020年第27号). 国家市场监督管理总局,2020-04-07.
[4] 《已上市化学药品变更事项及申报资料要求》 (2021年第15号).国家药品监督管理局,2021-02-10.