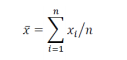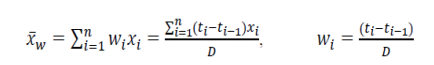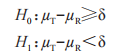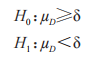摘要
美国食品药品管理局(FDA)于2023年4月发布了“简化新药申请的透皮和局部给药系统(TDS)黏附性评估的供企业用指导原则草案”,修订了2018年公布的同名指导原则草案。该修订的指导原则草案包括前言、背景、黏附性的评价、黏附性和生物等效性的综合评价以及数据提交格式5部分。重点讨论了仿制TDS黏附性的临床评价,包括其研究设计和实施以及统计分析。中国目前还没有类似的指导原则,详细介绍FDA的该指导原则草案,期望对我国仿制的TDS的黏附性评估的临床研究和药品监管部门的审评有帮助。
关键词:美国食品药品管理局(FDA);简化新药申请;透皮和局部给药系统;黏附性;临床评价;指导原则。
美国食品药品管理局(FDA)在 2023 年 4 月发布了“简化新药申请的透皮和局部给药系统黏附性评估的供企业用指导原则草案”(修订版2)[1]。该指导原则草案由FDA的药物评价与研究中心(CDER)仿制药办公室的研究和标准办公室与CDER的新药办公室和药品质量办公室合作编写。该指导原则草案(修订版2)修订了2018年10月10日在“联邦公报”上公布的同名指导原则草案修订版1[2]。该修订的指导原则草案对评估透皮或局部给药系统(TDS)黏附性能的研究设计和实施,提出了建议。根据仿制 TDS 产品开发方案的目的,申请人可以选择,在仅仅评价TDS黏附性的研究中,或者在具有综合目的的研究中评价 TDS 黏附性[例如,同时评价黏附性和生物等效性(BE)以及药动学(PK)终点]。该修订的指导原则草案中的建议,涉及为支持简化新药申请(ANDA)而提交的研究资料。
我国目前还没有类似的指导原则,本文详细介绍 FDA 该指导原则的修订版 2,期望对我国仿制的透皮和局部给药系统的黏附性评估的临床研究和药品监管部门的审评有帮助。
一、该指导原则的前言
该指导原则为评价 TDS 黏附性能的研究设计和实施,提出了建议。TDS包括可能在其他地方描述或称为贴片(patches)、局部贴片(topical patches)或缓释膜(extended-release films)的产品。该指导原则中的建议涉及,为支持 ANDA 提交的研究资料。根据仿制TDS产品开发方案的目的,申请人可以选择,在仅仅评价 TDS 黏附性的研究中,或者在具有综合目的的研究中评价 TDS 黏附性(例如,同时评价黏附性和BE以及PK终点)。
在新药申请或补充新药申请中表征 TDS 黏附性的研究建议,可能与支持 ANDA 的建议不同,并且可能涉及可能给予不同解剖部位的 TDS 产品的不同使用年龄和规格的评估。此外,支持新药申请研究的TDS黏附性的设计、实施和评估与其有本质的差别,因为在这种情况下,TDS 黏附性通常不与参比制剂对比评价。有关更多详细信息 ,请参阅“新药申请提交的局部和透皮系统黏附性评估”供企业用的指导原则草案(2021年7月)[3]。最终,该指导原则将代表FDA目前对这一主题的看法。FDA定期更新指导原则。有关最新版本的指导原则,请查看FDA 指导原则网页 https://www. fda. gov/regulatory information/search-fdaguidance-documents.
在该指导原则中,字母T[代表试验(Test)]将指作为 ANDA 对象的拟议的仿制药,字母 R[代表参照(reference)]将指参比制剂(RLD)和(或)标准制剂(reference standard product)。FDA 建议申请人在考虑设计和实施可能适合支持拟用仿制 TDS 产品与其 RLD 生物等效性对比研究时,参考该指导原则以及任何相关的特定产品指导原则(PSG)[4]和任何相关的企业用的指导原则[5-6]。FDA还建议申请人,定期访问 FDA的网站,因为可能有其他的指导原则,有助于开发仿制 TDS产品。
FDA鼓励,申请人为设计和进行评价TDS黏附性能的研究,寻求使用相关特定产品指导原则中FDA 建议的替代方法,并请联系该机构,讨论评价该特定药品黏附性能的拟议的替代方法[7-9]。
该指导原则修订了2018年10月发布的“ANDA透皮和局部给药系统黏附性评估供企业用的指导原则草案”[2]。这次修订阐明了测量黏附在皮肤上的整个 TDS 表面积估计百分比的适当方法以及该数据的统计分析。还鼓励申请人探索使用替代量表(并非第“3.1”节,研究设计和实施中,描述的 5点黏附性量表),估计 TDS 对皮肤的黏附性 ,并在ANDA 前会议上与 FDA 讨论这些替代量表[7-9]。此外,这次修订阐明了目前不打算使用照片证据,自动或光度分析,然而可以用于支持对每个时间点报告的黏附百分比的视觉观察。
一般来说,FDA的指导文件并没有规定法律上可强制执行的责任。相反,指导原则描述了该机构目前对某一主题的看法,除非引用了具体的监管或法定要求,否则仅应视为建议。在该机构指导原则中使用“should”一词意味着建议或推荐某些事物,而不是规定。
二、该指导原则发布的背景
通过 TDS 释放到患者皮肤并穿过患者皮肤的药物量在一定程度上取决于给药的表面积。在RLD说明书中包含的使用条件下,TDS的全部接触表面积应在整个敷用过程中,始终均匀地黏附在患者的皮肤上。如果TDS在敷用过程中失去黏附性,释放给患者的药物量可能减少。
在RLD的说明书的敷用期内,预计TDS可遇到由身体运动引起的扭转应变;环境温度或湿度的变化,例如每天暴露在水中(如在常规淋浴期间);以及与衣物、床上用品或其他表面接触。在RLD说明书的敷用期间,不能与皮肤保持一致和均匀黏附的TDS 产品,在产品敷用期间的不同时间,可能经历不同程度的TDS脱离,包括完全脱离。
如果TDS的黏附特性不够牢固,当根据RLD说明书的使用条件评价时,TDS可能在与皮肤接触的表面积方面存在不确定性。如,当 TDS 部分脱离时,产生的药物释放特性可能存在不确定性,因此,从 TDS 吸收药物的速率和程度也可能存在不确定性,这可能影响FDA对拟议仿制药的生物等效性评价。当TDS完全脱离的可能性增加时,药物产品意外暴露于意外接受者(如,可能是儿童的家庭成员)的风险也可增加。
三、黏附性的评价
3.1 研究设计和实施
一般来说,FDA建议申请人设计他们的黏附性研究,以支持对T和R TDS黏附特性的比较评价。
FDA建议申请人使用单剂量、随机、两治疗、两周期的交叉研究设计,所有受试者都敷用相同规格的 T 和 R TDS。然而,如果申请人适当地证明研究设计的合理性,FDA 也可以考虑使用单周期、每个受试者两治疗设计的研究(如配对研究)的可接受性,并且敷用部位随机化。TDS黏附性研究的人群通常应与产品 PK BE 研究中纳入或建议纳入的人群相同,通常应包括健康男性和未妊娠、未哺乳的女性,除非对产品具体考虑,另有说明。
申请人应在规定的研究周期,将受试者随机分到接受 T 或 R TDS 产品。在可能的情况下,第 2 个研究周期给予的 TDS,应当用于与第 1 个研究周期相同的解剖部位,但应选身体的对侧。
由于产品设计、活性或非活性成分、背衬膜或生产工艺的改变,可影响 TDS 的黏附特性,因此研究应使用即将上市的 TDS 产品。TDS 的批准后变更,可能需要确认与黏附性相关的产品质量特性,与TDS产品特征的产品质量特性保持一致,TDS产品在最初的ANDA批准中,已被证明有可接受的黏附性。
除非另有说明,否则在进行黏附性研究时,申请人应使用适用的特定产品指导原则中建议的TDS 的特定尺寸或规格。较大的 TDS 可能比较小的TDS更易脱离,因为较大的TDS可受到更大的构象或扭转应变,这些应变是由潜在增加的解剖曲率引起的,或者是由较大的TDS可黏附的相对较大的解剖距离上的更大幅度的屈曲引起的。申请人用较大的 TDS 比用较小的 TDS 更准确地评估黏附性评分,也是可能的。申请人不应因为盲法,而使用涂层或覆盖物,因为涂层或遮盖物可影响产品的作用。
申请人应在敷用TDS后的多个时间点,评价每个 TDS 的黏附性,以提供足够的分辨时间,从而在整个敷用期间充分比较 T 和 R TDS 的黏附特性。如,具有7 d敷用期的TDS的黏附性,应至少每天在相等间隔的时间点(如 24、48、72、96、120、144、168 h)评估;具有 72 h 敷用期的 TDS 的黏附性,应至少每12小时(如12、24、36、48、60、72 h)评估1次;敷用期在 12~24 h 的 TDS 的黏附性,应至少每 4 小时评估1次;以及敷用期小于12 h的TDS的黏附性,应当至少每小时评估1次。
此外,申请人通常应以均匀的方式分布这些时间点,在整个 RLD 说明书的敷用期内等距分布,因为根据个体评估计算的平均黏附性评分,旨在代表整个敷用期。对于某些 TDS,敷用早期的黏附性可能比敷用后期更好;因此在TDS敷用期早期较多的黏附性评估,可能(1)在 TDS 黏附性可能相对较好的初始阶段,通过过度表示黏附性评估,对平均黏附性评分的计算,不成比例的加权;(2)以不能代表该TDS的整个敷用持续时间的方式,不适当地降低平均黏附性评分。申请人应根据本节后面所述的建议,计算平均黏附性评分。在记录TDS黏附性的测量值时,申请人可以使用适当的方法[如经过培训的视觉评估和(或)点阵模板]和替代量表(下述的5点黏附性量表除外),估计黏附皮肤上的整个 TDS 表面积的百分比。如果申请人使用不同于下述 5点黏附性量表(表 1)的量表 ,记录 TDS 黏附性测量值 ,则他们应该将每个TDS 黏附性测量结果报告为,根据所选量表的评分,以及根据5点黏附性量表的相应评分。例如,如果观察者根据5点量表,将TDS黏附性评为2分,并估计产品似乎黏附了 60%,则应报告该时间点的评分为2分和其估计值为60%。可以考虑基于替代量表评分的信息和(或)分析,前提是替代量表的使用是合理的,并且该信息与研究资料一起提交,以证明该量表足以符合要求。
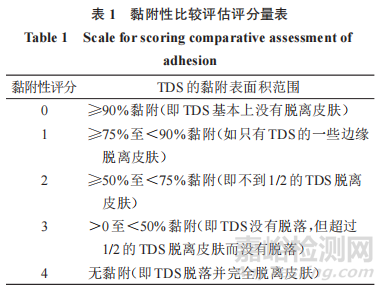
对于黏附性的比较评估,即第 3.2 节中描述的非劣效性(NI)试验,申请人应使用下列 5 点黏附性量表,其中每个评分对应于TDS的特定黏附表面积范围。
在每个黏附性评估时间点,申请人还应记录,显示 TDS 与皮肤黏附程度的照片证据。由于黏附百分比可包括 1 个范围,但可以归类为 1 个单独的评分,因此照片证据可用于支持对每个时间点报告的黏附百分比的视觉观察,但此时不拟用于自动或光度分析。
对于每个时间点的每次连续TDS黏附性测量,申请人应根据该时间点的 TDS 黏附性实际测量结果记录评分(不结转前一时间点的评分),无论分数相对于前一评分是增加还是减少。连续的 TDS 黏附性测量应独立于先前的测量,并且观察者对先前的测量处于盲态。
然而,在分析黏附性比较评估的结果(即第 3.2节中描述的 NI 试验)时,在基线或时间 0 之后的任何时间点,使用上述 5 点黏附性量表评估的最高黏附性评分(即表示该 TDS 的最大脱离程度的评分)应用于后续时间点,直到评估出更高的评分。例如,如果黏附性评分是1、2、1、3,那么估算的黏附性评分将是 1、2、2、3。对于完全脱离的 TDS,在整个研究期间 ,应为该 TDS 方案的任何剩余评估 ,评为4分。
申请人应当使用平均黏附性评分( 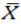 )作为评价TDS 黏附性的主要终点。对于 TDS,应根据其在每个评估时间点的个体黏附性评分得出,在所有等距离时间点(基线时间点 t0,除外)取平均值。由表示在基线之后所有 n 等距离的时间点,观察到的TDS的平均黏附性评分。可如下计算:
)作为评价TDS 黏附性的主要终点。对于 TDS,应根据其在每个评估时间点的个体黏附性评分得出,在所有等距离时间点(基线时间点 t0,除外)取平均值。由表示在基线之后所有 n 等距离的时间点,观察到的TDS的平均黏附性评分。可如下计算:
在这里,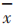 是在基线后等距时间点上,观察到的TDS的平均黏附性评分,以及
是在基线后等距时间点上,观察到的TDS的平均黏附性评分,以及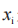 是在第i次测量时观察到的黏附性评分,或如前所述的 TDS 直到第 i 次检测为止的最高观察评分。
是在第i次测量时观察到的黏附性评分,或如前所述的 TDS 直到第 i 次检测为止的最高观察评分。
尽管该指导原则中的建议是以均匀、等距的方式分配时间点,但如果数据集包含来自非等距时间点的评分,则加权平均值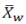 (具有与区间长度相对应的权重)可以如下计算:
(具有与区间长度相对应的权重)可以如下计算:
在这里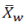 是在基线之后所有非 n等距时间点,观察到的TDS的加权平均黏附性评分;
是在基线之后所有非 n等距时间点,观察到的TDS的加权平均黏附性评分;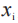 是第i次测量时,观察到的黏附性评分;
是第i次测量时,观察到的黏附性评分;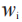 是
是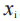 对应的权重;D是敷药的总持续时间;
对应的权重;D是敷药的总持续时间;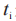 是第 i个测量时间;和
是第 i个测量时间;和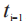 是前 1个(i-1)测量时间。由于计算机软件可能存在舍入误差,FDA建议申请人首先计算分子中的总和,然后将该总
是前 1个(i-1)测量时间。由于计算机软件可能存在舍入误差,FDA建议申请人首先计算分子中的总和,然后将该总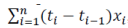 和除以总持续时间D。
和除以总持续时间D。
例如,对于 24 h 敷用 TDS,如果申请人在基线后第 2、4、8、12、24 h 测量黏附性,则敷用的总持续时间将为 24 h。系数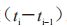 对应于第 i 次测量
对应于第 i 次测量 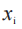 (i=1,2,3,4,5)分 别 为(2-0)、(4-2)、(8-4)、(12-8)和(24-12)。首先,加权平均值
(i=1,2,3,4,5)分 别 为(2-0)、(4-2)、(8-4)、(12-8)和(24-12)。首先,加权平均值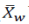 可通过求和
可通过求和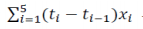 计算,然后将总和除以总持
计算,然后将总和除以总持
续时间 D(本例中为 24 h)。所有 5个测量值的相应权重为1/12、1/12、1/6和1/2,加起来为1。
除了主要终点外,FDA 建议申请人使用上述5点黏附性量表,进行下列描述性分析,评价 TDS 黏附性,从而评估治疗组的潜在临床意义价值或事件的差异:(1)在任何时间点观察到的黏附性评分≥2的受试者比例,在 T 和 R 之间比较。(2)R 平均黏附
性评分,比相应的 T 平均黏附性评分大 1 或更多的受试者的比例,与其T平均黏附性评分,比相应的R平均黏附性评分大 1 或更多的受试者的比例相比。(3)T和R之间观察到的黏附性评分≥2的时间。如果有足够数量的事件,可绘制 Kaplan-Meier 累积发生率曲线。
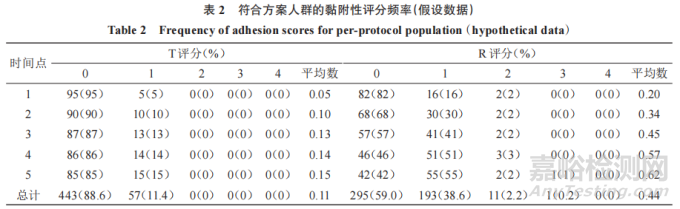
此外,申请人应在频率表中提交描述性黏附性评分数据,说明在每个评估时间点和所有时间点的每个黏附性评分的 T和 R TDS的数量和比例。表2是频率表的样式,以n=100计算T评分、R评分。
申请人应注意,应以 RLD 说明书所述的方式,给研究受试者 T和 R TDS,并应在 RLD的最大说明书持续时间内,评估 TDS 黏附性。一般来说,研究期间不应限制研究受试者的活动;相反,应允许受试者在研究单位内和(或)在家中自由进行正常活动(如进行淋浴等真实世界的活动),这些活动可能合理地预计在产品的说明书使用期限内发生。对于敷用时间长达或超过24 h的产品,FDA建议受试者在研究期间,可以按照 RLD 的说明书使用方式,常规沐浴或淋浴,并且在此类常规活动中,不应保护TDS不直接暴露于水。
通常,申请人应仅使用完整无损的T和R TDS,评估相对黏附性,因为改变 TDS 的大小或形状,可能改变其黏附特性。申请人应在其研究方案中包括相关规定,以确保在整个研究过程中避免故意采取行动,重新敷于TDS 的脱离区域,对 TDS 施加压力,或加强 TDS 与皮肤的黏附(如覆盖物)。研究方案应包括确保TDS脱离不受到不适当限制的规定(如通过座椅靠背对TDS的持续施压)。受试者不应在放置TDS的皮肤区域,涂抹化妆品、面霜、乳液、粉末或其他局部产品,因为它们可能影响黏附性能。此外 ,在敷用 TDS 之前 ,应剪掉(而不是剃掉)敷用部位的毛发,并且应以与RLD的说明书使用一致的方式,准备敷用部位。申请人应在研究方案中描述随机化方法,并以XPT 格式的 SAS 传输数据集,提供随机化方案(注意 ,本文中的随机化指的是序列 ,而不是治疗)。FDA建议在整个研究过程中,由独立的第三方生成并保存随机化代码,以最大限度地减少偏倚。然而,如果申请人没有参与研究药物的包装和标示,那么其生成随机代码可能是合适的。申请人应确保随机方案的密封副本保留在研究现场,并且该密封副本应在现场检查时 ,提供给 FDA 的调查人员,以便验证每个受试者每个敷用部位的治疗身份。
3.2 统计分析的考虑
申请人应预先规定用于黏附性分析的符合方案(PP)人群,并根据每个受试者的TDS定义。用于黏附性分析的PP人群应包括所有TDS,但在研究早期故意去除的TDS除外(如由于不可接受的刺激),或在RLD说明书的敷用时间结束前,因与黏附性无关的原因(如因为违反方案),停止使用TDS的受试者的TDS除外。申请人应在其研究报告中包括,描述被排除在PP人群之外的任何受试者的个例报告,以及排除受试者的原因。
申请人应比较T和R产品的每个治疗组的平均黏附评分(即上述主要终点)的平均数。为了计算平均黏附性评分,申请人应将基线时间点(t0)后每个时间点的最高黏附性评分,结转到后续时间点,直到评估出更高的评分。为了证明足够的产品黏附性,申请人应根据评价 T 和 R 群体平均黏附性评分差值,证明 T 产品在统学上不劣效于 R 产品,NI界值为 0.15(δ=0.15)。0.15 的 NI 界值适用于基于先前描述的 5点黏附性量表的 T和 R产品的平均黏附性评分差值;0.15的NI界值不适用于基于其他黏附性量表或非基于位置的数据转换(如对数转换)的平均黏附性评分差值,也不适用于 T 和 R 的黏附性评分中位数差值。
申请人应在0.05 的显著性水平,检验下列假设:
在这里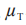 和
和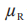 分别是 T 和 R 产品平均黏附性评分的群体平均数,以及替代假设 H1表示 T产品的黏附性相对于 R 产品的黏附性的 NI。这些假设对应于下列内容:
分别是 T 和 R 产品平均黏附性评分的群体平均数,以及替代假设 H1表示 T产品的黏附性相对于 R 产品的黏附性的 NI。这些假设对应于下列内容:
在这里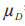 等于T和R产品的平均黏附性评分的群体平均数差值:
等于T和R产品的平均黏附性评分的群体平均数差值: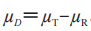 。当在交叉或配对研究中没有丢失数据时,
。当在交叉或配对研究中没有丢失数据时,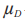 与在配对T(
与在配对T(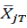 )和R(
)和R(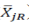 )个体受试者 j的平均黏附性评分差值的群体平均数
)个体受试者 j的平均黏附性评分差值的群体平均数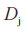 相同]
相同]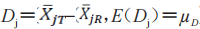 。
。
为了证明 T 产品的可接受的黏附性,申请人应设计并进行如上所述的黏附性研究,并招募足够数量的受试者,以使研究达到 0.80或更高的水平。由于黏附量表的离散性和黏附数据的其他潜在复杂性,FDA 建议申请人使用足够大的样本量,以确保任何大样本(渐进)高斯(Gaussian)假设的有效性(如果使用的话)。
不完整的数据和与不依从相关的数据可能损害NI研究的有效性 。FDA建议良好的临床研究设计和实施,以防止受试者退出和不依从。尽管如此 ,当 这些事件发发生时。申请人应记录这些事件的详细原因。尽管FDA建议将PP人群作 为 NI 研究的主要分析人群 ,但 FDA也对信息的丢失和不合规的可能性表示特别担忧。如果适用 ,申请人应在其方案中预先规定插补方法。FDA建议申请人预先规定敏感性分析,以评估任何不平衡或信息缺失和不合规,对黏附性NI结论的潜在影响。
四、黏附性和BE的综合评价
如果申请人选择在单一研究中,进行评价T和R产品的黏附性能和 PK BE 的研究,这种研究应在足够大的人群中进行,以充分支持黏附性的比较评价,并包括足够大的受试者亚群,以适当选择 PK终点,充分支持 BE 的评价。申请人应根据方案中预先规定的计划,选择PK BE评价的参与者。
第“3.1”节中所述的研究设计和实施建议,针对专门为评价TDS黏附性而进行的研究,也适用于评价黏附性和具有PK终点的BE联合研究。
在TDS黏附性和PK BE的联合研究中,对受试者同时敷用多个 T TDS 或多个 R TDS 可能是合适的,如果这样做是安全和合理的,可能需要增加药物释放,以补偿测量 PK 样品中相关分析物的分析灵敏度不足。在这种情况下,当多个TDS同时应用于受试者时,应评估每个和所有TDS的黏附性能。
申请人应收集和分析 PK亚群中所有受试者的PK样品,而不考虑受试者 TDS黏附性评分,并报告PK 研究中所有时间点的样品浓度以及所有受试者的 PK 结果。体内黏附性和(或)PK BE研究结束时移除的(或在研究过程中脱离的)所有 TDS 单位应保留,用于分析残留药物含量[10-11]。
申请人应预先规定 PK终点统计分析的纳入标准,并对相关人群(可能是PP人群的子集)进行初步PK 分析。FDA 还建议申请人查阅关于PK分析BE标准建议的相关特定产品的指导原则。
五、数据提交格式
申请人应以标准化格式提交研究数据,并参考FDA关于提交给 CDER的研究数据的网页[12],了解有关研究数据标准的更多信息。此外,申请人应提供XPT格式的SAS传输数据集和定义文件。如果采用插补,申请人应提交插补后的原始数据和分析数据。
六、结语
FDA的“简化新药申请的透皮和局部给药系统黏附性评估的供企业用的指导原则草案(修订版2)”,重点讨论了仿制 TDS(不是新药申请的 TDS)黏附性的临床评价,包括其研究设计和实施以及统计分析。这次的修订版与2018年修订版相比,阐明了测量黏附在皮肤上的整个 TDS 表面积估计百分比的方法以及该数据的统计分析。
6.1 该指导原则草案(修订版2)中值得关注的内容
FDA的“简化新药申请的透皮和局部给药系统黏附性评估的供企业用的指导原则草案(修订版2)”详细而具体地描述了仿制TDS的临床研究方案及其实施方法。该指导原则对中国这方面的临床研究和药品监管部门的审评有很好的参考价值。其中值得特别关注的包括但不限于下列6点内容:
(1)FDA建议仿制TDS黏附性评估研究采用单剂量、随机、两治疗、两周期的交叉研究设计。
(2)研究应使用即将上市的 TDS 产品;应使用适用的特定产品指导原则中建议的 TDS 特定尺寸或规格。
(3)应在敷用 TDS 后的多个时间点,评价每个TDS 的黏附性;在每个黏附性评估时间点,还应记录TDS与皮肤黏附程度的照片证据。
(4)黏附性比较评估的5点黏附性量表。
(5)应以 RLD 说明书所述的方式,给研究受试者 T和 R TDS,并应在 RLD的最大说明书持续时间内,评估TDS黏附性。
(6)申请人应根据评价 T 和 R 群体平均黏附性评分差值,证明T产品在统学上不劣效于R产品,NI界值为0.15(δ=0.15)。
6.2 中国应制定仿制 TDS 黏附性评估临床研究指导原则
通过 TDS 释放到患者皮肤并穿过患者皮肤的药物量在一定程度上取决于给药的表面积。如果TDS在敷用过程中失去黏附性,释放给患者的药物量可能减少。为此,FDA 发布了“简化新药申请的透皮和局部给药系统黏附性评估的供企业用的指导原则草案(修订版2)”。
而中国目前还没有类似的指导原则,这将影响我国对仿制TDS安全有效性的评价。2020年版《中国药典》四部的通则中第 0952 条“黏附力测定法”,只提供了体外的测定方法,而没有人体内的黏附性测定方法[13]。国家药品监督管理局药品审评中心2020年12月发布的“化学仿制药透皮贴剂药学研究技术指导原则”[14],在“体外黏附性能”一节中,也没有提及人体内黏附性的测定。国家药品监督管理局药品审评中心 2021年 3月发布的“皮肤外用化学仿制药研究技术指导原则(试行)”[15]提出了对这类仿制药开发过程中药学研究、非临床研究和生物等效性研究的技术要求,但没有涉及人体内的黏附性研究要求。国家药品监督管理局药品审评中心2022 年 5 月发布的“局部给药局部起效药物临床试验技术指导原则”[16]也未涉及人体内的黏附性研究。建议参考本文介绍的FDA指导原则,结合我国实际,制定仿制 TDS 人体黏附性研究的技术指导原则 ,规范这方面的研究和药品监管部门的审评。
参考文献
[1] FDA. Assessing Adhesion With Transdermal and Topical Delivery Systems for ANDAs Draft Guidance for Industry [EB/OL]. (2023-04-13) [2023-05-18].https://www.fda.gov/media/167043/download.
[2] FDA. Assessing Adhesion With Transdermal and Topical Delivery 2 Systems for ANDAs Draft Guidance for Industry [EB/OL]. (2018-10-10) [2023-05-18]. https://www.fda.gov/media/98634/download.
[3] FDA. Assessment of Adhesion for Topical and Transdermal Systems Submitted in New Drug Applications Draft Guidance for Industry [EB/OL].(2021-07-01) [2023-05-18]. https://www. fda. gov/media/150509/download.
[4] FDA. Product-Specific Guidance for Generic Drug Development [EB/OL]. (2022-12-11)[2023-05-18]. https: //www. fda. gov/drugs/guidances-drugs/product-specific guidances-generic-drug-development.
[5] FDA. Assessing the Irritation and Sensitization Potential of Generic Transdermal and Topical Delivery Systems for ANDAs Draft Guidance for Industry1 [EB/OL]. (2023-04-13) [2023-05-18]. https://www. fda. gov/media167073/download.
[6] FDA. Transdermal and Topical Delivery Systems —Product Development and Quality Considerations Draft Guidance for Industry [EB/OL]. (2019-11-21) [2023-05-18]. https://www.fda.gov/media/132674/download.
[7] FDA. Manual of Policies and Procedures (MAPP) 5220.8 Evaluating Requests for and Conducting Product Development and Pre-Submission Pre-ANDA Meetings [EB/OL]. (2022-10-05) [2023-05-18]. https://www. fda.gov/media/130874/download.
[8] FDA. Controlled Correspondence Related to Generic Drug Development Guidance for Industry [EB/OL].(2022-12-21) [2023-05-18]. https://www. fda. gov/media/164111/download.
[9] FDA. Meetings Between FDA and ANDA Applicants ofComplex Products Under GDUFA Guidance for Industry [EB/OL]. (2022-10-05) [2023-05-18]. https://www.fda.gov/media/107626/download.
[10] FDA. Residual Drug in Transdermal and Related Drug Delivery Systems Guidance for Industry [EB/OL]. (2011-08-17) [2023-05-18]. https://www. fda. gov/media/79401/download.
[11] FDA. Ransdermal and Topical Delivery Systems —Product Development and Quality Considerations Draft Guidance for Industry [EB/OL]. (2019-11-21) [2023-05-18]. https://www.fda.gov/media/132674/download.
[12] FDA. Study Data for Submission to CDER [EB/OL].(2018-06-06) [2023-05-18]. https://www. fda. gov/drugs/electronic-regulatory-submission-and-review/study-data submission-cder.
[13] 中国药典 [S]. 四部. 2020.Pharmacopoeia of the People's Republic of China [S].Volume IV. 2020.
[14] 国家药品监督管理局药品审评中心 . 化学仿制药透皮贴剂药学研究技术指导原则 [EB/OL]. (2020-12-25)[2023-05-18]. https://www. cde. org. cn/zdyz/domesticinfopage? zdyzIdCODE=2896eacb6c9b84ef3ec6f6b01b 7531e2. Center for Drug Evaluation, NMPA. Guidance for pharmaceutical research technology of chemical generic transdermal patches [EB/OL]. (2020-12-25)[2023-05-18].
https://www. cde. org. cn/zdyz/domesticinfopage?zdyz IdCODE=2896eacb6c9b84ef3ec6f6b01b7531e2.
[15] 国家药品监督管理局药品审评中心 . 皮肤外用化学仿
制药研究技术指导原则(试行) [EB/OL]. (2021-03-03)[2023-05-18]. https://www. cde. org. cn/main/news/ viewInfoCommon/4e790f4ad1cb21091e5a8bf4a107c535.Center for Drug Evaluation, NMPA. Guidance for the Research Technology of Chemical Genetics for Skin External Use (Trial) [EB/OL]. (2021-03-03) [2023-05-18]. https://www. cde. org. cn/main/news/viewInfoCommon/4e790f4ad1cb21091e5a8bf4a107c535.
[16] 国家药品监督管理局药品审评中心 . 局部给药局部起效药物临床试验技术指导原则 [EB/OL]. (2022-05-30)[2023-05-18]. https://www. cde. org. cn/main/fullsearch/fullsearchpage.Center for Drug Evaluation, NMPA. Guidance for Clinical Trial Techniques of Locally Administered andLocally Effective Drugs [EB/OL]. (2022-05-30)[2023-05-18]. https://www. cde. org. cn/main/news/viewInfo Common/f993bea8924aff71b361ad907612dbcd.