随着人用药品技术要求国际协调理事会(ICH)Q13指南的发布以及多个采用连续制造技术的口服固体制剂获批,药品连续制造在该类产品领域的监管发展受到越来越多的关注。物料处理是连续制造平台重要的组成部分,本研究分析了物料输入、连续给料和连续混合方面的技术特点,介绍了口服固体制剂连续制造的物料处理步骤,并从工艺验证、稳健性、设施设备、过程控制、质量保证和清洁等方面探讨了监管检查的关注点。在相关技术考量方面,工艺设备应能持续稳定地传递物料且尽可能控制风险并降低物料损耗,因此需要深入了解物料特性,科学地设计工艺设备和开展确认验证;在监管检查方面可关注连续工艺的专属风险以及对产品质量可能产生的影响。期望能以此促进连续制造在制药行业的应用,同时也建议监管机构、制药企业以及全社会共同参与,充分运用过程分析技术、创新检验方法和数字化质量保证等手段助力持续生产出符合质量要求的产品,从而推动我国制药工业向高水平现代化迈进。
随着 2021 年 7 月人用药品技术要求国际协调理事会 (The International Council for Harmonisationof Technical Requirements for Pharmaceuticals forHuman Use,ICH)Q13《原料药和制剂的连续制造》指南发布征求意见[1],药品连续制造 (pharmaceuticalcontinuous manufacturing,PCM) 的监管发展进入了新的时代。全球主要监管机构一致鼓励连续制造技术的应用,2015 至 2021 年,美国、欧盟以及日本陆续批准了涉及 3 类工艺生产线、4 家公司的 9个口服固体制剂 (oral solid dosage,OSD) 上市 [2-3]。总体上,药品连续制造在全球应用呈增长趋势,近年来该领域科学技术的快速发展促进了全球业界实践以及监管经验的积累,对我国药品连续制造的发展也起到了持续推动作用。
从全球监管考量来看,美国食品药品监督管理局 (FDA)、日本独立行政法人医药品医疗器械综合机构 (PMDA) 等也都发布有药品连续制造相关的指南细则 [4-7];欧洲药品管理局 (EMA) 虽然暂没有提供具体指南,但已通过多种场合表示其目前的监管体系足以支持连续制造的应用 [8]。监管考量具有一定的地区特点,相对来说,ICH Q13 总体还是比较框架性的,其延伸更宽泛、包容度更高、在申报资料指导方面更详细 ;FDA、PMDA 指南相对更具体,在专业领域的聚焦较深、较细 [9]。除监管机构外,其他机构或组织也在积极推进先进制造。美国药典委员会在药品连续制造的控制策略标准化、成分表征标准化 ( 体现在文献标准和实物参比标准 )、产品放行标准化、设备 / 系统要求标准化、传感技术标准化以及系统建模标准化方面一直在开展深入研究 [10] ;美国材料与试验协会 (ASTM) 于2014 年发布了“制药工业连续工艺应用的标准指南”[11] ;国际制药工程协会 (ISPE) 口服固体实践委员会连续制造分委会 2022 年也发布了“良好实践指南 :口服固体制剂的连续制造”[12]。这些成果都在推动连续制造创新和变革过程中发挥了重要作用。各指南的侧重点简要对比见表1。
表1 连续制造相关指南的对比
口服固体制剂连续制造已有较为成功的经验,当前阶段的相关探讨也较多聚焦在该领域。物料处理是连续制造平台重要的一部分,应采用合适的设备,使用特性良好的物料,并采取良好的过程控制,使中间品及成品质量始终在其关键质量属性(critical quality attribute,CQA) 受控状态范围内。口服固体制剂生产中,可能涉及液体加工及干燥颗粒加工时的物料特性表征与控制,而物料特性对系统的整体行为起重要的作用,在一定程度上,物料的可变性很可能是影响产品质量的主因,因此充分理解物料性能、合理选取和设计设备,在工艺中通过过程分析技术 (process analysis technology,PAT)监控和反馈,动态维持受控状态,更符合质量源于设计 (Quality by Design,QbD) 理念。在 ICH、FDA 指南的基础上,ISPE 指南从制药工程的角度纳入了口服固体制剂的业界共识,是对监管指南的重要补充。本研究重点参考 ISPE 指南的部分内容,讨论在口服固体制剂的连续制造过程中持续稳定处理物料的相关考量,探讨监管检查可能需要关注的要点。
1、口服固体制剂连续制造的物料处理
药品连续制造的物料处理应尽量在不引入工艺风险和不造成物料损失的情况下,持续稳定地将物料在连续制造设备系统中进行传递。这就需要在了解物料特性的基础上确定适合的设备类型和属性,并基于科学设计和确认验证数据来建立基本工艺描述和最低性能指标要求。考虑到连续制造的“批次”运行时间较长、物料处理量较大,且生产中途有可能出现扰动等情况,还应关注操作安全 ( 如粉尘爆炸风险控制等 )、产品切换、可清洁性以及组装和拆卸便利性等 [12]。
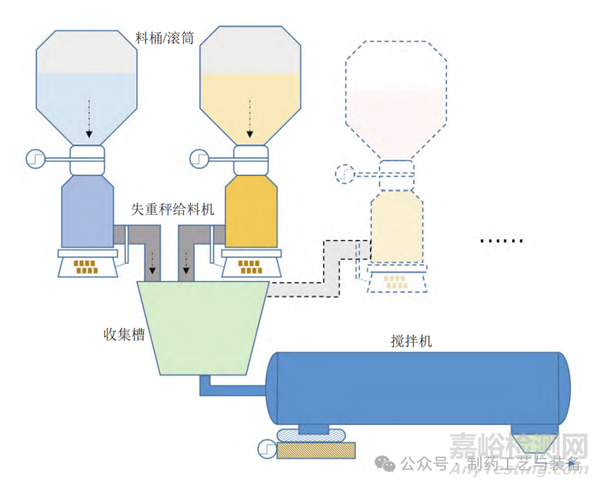
物料处理过程一般包括物料的存储、连续给料、混合前收集、搅拌混合以及过程中的各类输送环节等。多数口服固体制剂尚未能实现端到端连续,一般通过中间料斗在适当批量和时间内保持工艺连续。不同粉末原料的物理性质可能不同,对前序工艺条件的保持能力也有差异,因此需要综合考虑采用机械助流等主动方式驱动粉末流动或通过重力排出等被动方式引导流动的设备设计。连续工艺的前端涉及原料、物料或中间品的投入,需要主动计量并对投入的物料流进行混合 ;为达到所需的精细控制水平,常需采用批量工艺和连续工艺配合操作。初始物料输入通常为批量给料设备,一旦物料进入生产线,则一般采用失重秤给料机。常见的 PCM物料处理的设备和流程如图 1 所示。
图1 药品连续制造的物料处理示意图
1.1物料输入
粉末处理不仅是物料输入的重点,也是多数连续制造单元操作中的关注点。粉末的特征影响操作性能,决定了设备和工艺参数的选取及确认。粉末处理特性概况可参见表 2 中的描述 [12]。在物料输入工艺的研发和试制过程中应充分考虑这些特性对工艺和产品的影响,合理设计、验证并制定有效的管理措施。
表2 粉末特性的影响 1)
例如,有黏性和可充气的物料难以自由流动和快速沉降,需要考虑辅助输送 ;采用稀相输送流速快且不易堵塞,但可能会导致管道或颗粒磨损及偏析的风险,此外,稀相输送不适于静电敏感性物料,可能导致颗粒黏附在管道壁面 ;采用密相输送可降低颗粒磨损,但对物料要求较苛刻,例如用平推流(plug flow) 输送大颗粒和片剂,移动床流 (movingbed flow) 输送细的、不可渗透的粉末。正压输送系统可利用气压牵引粉末,而真空输送系统可在常压下将粉末吸入真空容器 ;正压输送距离远,而真空输送更适于处理易成粉或有毒物料。
在连续制造中,工序之间的连接点通常需要部署物料处理系统,以支持相邻工序的参数之间平稳转换或桥接。可靠的粉末流动能力是一个考虑重点,在开发、初始部署和变更时都应仔细评估。还应关注物料的粒径和流动性等性质。系统的性能取决于物料与设备特性之间的相互作用,在性能确认或试制之前,尽可能考虑建立基于 QbD 的物料处理模型和设计空间,重点研究进料和混合,并关注混合粉末的积累和处理的情况。
1.2连续给料
连续制造单元需具备一定的存储能力。未加工物料需要仓储,可能因自重或装袋堆积应力等导致凝聚性增加,影响初始流动性,需要结合物料的专属特性进行考量。例如,刘飞等分别采用连续干粉混合和直接压片制造缓释片,观察到原料的流动性对缓释片基质的可制造性和加工性有很大影响,具有良好流动性的粉末和有效的混合对于片剂质量稳定非常关键 [13]。
生产线的物料容器主要有料仓、滚筒和柔性袋等。料仓通常是不锈钢结构,优点是机械坚固、密封性好、易于清洗和反复使用 ;缺点是不灵活、难改造。滚筒通常是塑料衬里加不锈钢或纤维架构,灵活性好,可组合使用;但须注意滚筒和衬垫材料的潜在污染情况,以及较大滚筒中可能出现的较高顶压和沉降。柔性袋系统一般具有柔性、透明的外壁,可通过目视确认物料的排放情况 ;通常是一次性或即抛型的,减少了物料转移和清洁的需要 ;但在采用堆叠储存时,底部物料承受的累积质量可能导致粉末过度固结,影响流动性。实际应用中,需结合包装类型、库存需求以及实现方式确定总容量。此外,还须考虑应便于物料动态转移使用,以及大规格包装中剩余的物料存储评估等。
1.3连续混合
即便是单一原料发生偏析也会导致粗、细颗粒不均匀,从而影响下游操作的堆积密度及混合。对于 API 须与辅料预混合的情形,还存在拱起、拉孔、黏聚或结块、载气等潜在问题,即使质量流量相对恒定,仍可能导致物料在连续混合过程中随时间发生变异。因此,应采取有效手段分析物料混合情况,并维持连续混合物料的质量符合要求。例如美国罗格斯大学开发的连续直接压片系统,输入物料的质量可采用混合动力比例 - 积分 - 导数控制预测模型进行控制,按要求送入混合器形成均匀的混合物 ;德国 GEA 公司开发的商业型直接压片的ConsiGma-DC 型连续生产设备中则采用近红外光谱仪实时检测成分的混合均匀度 [14]。
在重力流不足以确保粉末可靠排出的情况下,需要考虑机械助流,包括全面助流或仅清空容器壁上的残留物料。典型方法包括外部振动、内部搅拌或振动、注气等。此外,原料可能需要通过选取合适的磨机类型如轧辊压缩、冲击或锤击、剪切或盘磨等进行研磨以消除结块、降低颗粒尺寸,以改善颗粒的粒度分布 (particle size distribution,PSD) 目标、形状和持续均匀性等。
2、监管检查关注点探讨
药品生产企业应采取有效的措施,确保采用药品连续制造工艺的口服固体制剂物料处理符合药品生产质量管理规范的要求。在监管检查过程中,除了关注批量生产中的一般要求之外,还需要重点关注连续制造物料处理的特殊之处及对产品质量影响的相应风险。
2.1工艺验证方面
部分单元操作在符合科学性的情况下可采用缩小模型验证;而部分工艺如外部振动机械助流,考虑到其拟解决的问题可能在小型测试平台中无法体现,需要在最大规模或接近最大规模条件下进行性能测试。监管检查可关注研发或生产企业开展工艺验证的策略和选取模型的合理性。
物料能够达到预期的传递过程是物料处理系统工艺验证关注的重要方面,需要评估输送类型和输送工具特点以及带来的相应影响。对于气体输送机这一类关键部件,监管检查可关注其能否提供所需的压力和流量,速度或物料类型可否允许调整,是否能基于压力与流量的性能曲线等选取规格,是否能经过充分验证来确保输送周期内给料速度的一致性。
此外,可关注企业的工艺验证是否充分考虑了管道连接点堆积、磨损和压力损失;关注企业所使用的分离器类型是否考虑了科学布局和适当的通量范围,选取过滤器表面清理方法的合理性,是否设置内部清洗程序,选择的过滤介质是否评估了操作温度、耐磨性和过滤性能等。
2.2工艺稳健性方面
物料处理系统须能长时间稳定运行,并与整个工艺流程相适应。生产期间应避免故障或性能损失,避免过量的噪声、振动、热积累等非预期情况,避免不适当的摩擦磨损或污染物产生。因此,应关注企业是否采取有效措施维持工艺稳健。
任何单元操作若存在粉尘,都须在防爆方面进行独立评估。研磨和筛分非常容易产生粉尘,应关注企业是否开展了粉尘危害分析,是否评估了可燃粉尘的标准化压力上升速率 (normalized rateof pressure rise,Kst)、最大爆炸超压 (maximumexplosion overpressure,Pmax)、最低可爆炸浓度、限制氧浓度、最小点火能量等的风险,是否在关键位置采取降速、惰性气体吹扫、接地、通风口和抑制系统等预防措施。
管道堵塞和滤料堆积可能对产品质量产生严重影响,监管检查可关注是否具备监测仪表,对粉末质量、气体压力、温度和气流等参数是否按需记录,是否采取反馈控制策略以降低相应的风险。如设计了管道清洗循环,可关注控制逻辑避免污染。关注生产线及其主要部件是否便于快速拆卸和内部检查或可更换磨损的部件,是否充分评估了维护和预防性检查的频率等。
在设备或单元间的连接方面,可关注灵活连接设备的刚性、可移动性、直接接触产品的材料、对齐情况等;采用固定溜槽连接时,要加入对其角度、横截面、内表面光滑度的考量,以防止堆积、确保物料流速稳定、减少黏附等。关注中间阀开口是否可防止积聚、扰动和泄漏,是否依赖传感器,是否可快速拆卸利于清洗等。
2.3设施设备方面
可关注进料槽或料斗容量能否维持转换期间的运行。小容器灵活但比大容器所需要的填充操作更频繁,检查时应关注选取存储设备时是否对针对工艺目标开展了评估。储罐的几何形状对流动性能影响较大,合适的料斗形状、光滑陡峭的内壁可以促进重力卸料时产生的质量流 ( 沿料斗壁流动 ),使物料先进先出、防止偏析。若物料以漏斗流的形式流动,则会导致潜在的流动障碍和难以清洁等问题,并造成排出物料堆积密度不稳定 ;料斗出口上方有物料积滞,导致出现物料先入后出的情况并增加偏析倾向。建议检查过程中关注设施设备是否考虑到其接触物料表面的材质、粗糙度、倾斜角度,并是否通过测试进行评估。
检查时还可关注流动辅助所需程度的计算是否合理,整个出口区域是否能够保持动态检查,阀门特性是否充分考虑了下游的响应时间、密封、空间、成本、维护和清洁。如果使用了连接和对接设备,可关注是否充分考虑到粉尘隔离、设备振动、接口尺寸和传输速率等因素。
2.4过程控制方面
过程控制可能需要采用系统组件如机械取样探头或接触点来收集样本,送至实验室离线分析物料的水分、粒径、含量等特性,监管检查时可关注样本的代表性,评估取样位置、设备大小和取样方法的合理性,以及是否考虑到流动床层干扰和取样区域限制等可能造成的偏差。采用 PAT 方式替代直接提取或去除离散粉末时,相关仪器须直接安装在工艺设备中以实时监测流经粉末的属性,检查时可关注采用的分析方法与所采集信息的相关性、设备位置、仪器有效测量区域的维持、校准情况以及采样率等。在全局取样计划方面,建议可以关注其在统计上是否合理,以及检测持续时间是否考虑到过程停留时间以便及时反馈信息并及时做出操作决策。
在口服固体制剂的连续制造过程中,很多不合格情况是由于物料或中间品的偏析造成的,应尽可能采用 PAT 等工具对工艺中不合格物料进行预防和监控。监管检查时可关注是否充分研究了颗粒体积、形状和密度,以及是否了解传输工序中的作用力和粒子间运动模式。特别应关注偏析可能的发生机制,如颗粒间筛分、空气夹带以及粉尘形成,是否采取了有效措施予以降低。
2.5质量保证方面
连续生产线性能与粉末成分及混合物的物理性能具有较强的相关性,不合格物料的产生很大程度上取决于工艺或设备组合、材料或配方特性和控制策略的设计。连续工艺不合格物料的发现、追溯和对产品质量的影响需要更深入分析,可能需要对物料性质、对应的表征方法以及停留时间分布(residence time distribution,RTD) 等方面进行深入研究才能进行科学评估。连续制造过程中的物料特性不一定是稳态的,但其动态变化应在验证的范围内进行整体考量,当发生较大偏离时,应考虑结合RTD 数据等科学依据对潜在的不合格物料加以分流 [15]。监管检查时可关注企业针对物料性质及工艺特点所制定的偏差调查规程和中间品、产品放行管理规程,并关注偏差调查的记录是否能反映出对原因的准确定位和风险的全面评估,以合理支持产品放行依据、提供充分的质量保证。
物料处理系统可能因共线生产需求而进行变更,例如,引入新产品时,各进料点需要能够适于处理不同特性和 (或) 使用率的物料。在产能需要扩大的情况下也可能会引起变更,例如复制生产线、增加单元操作设备、增加质量流量或延长运行时间等。监管检查时可关注企业质量保证体系是否对变更开展了有效管理,对于涉及不同于批制造生产模式的特定风险是否进行了充分评估,是否采取了充分的控制措施确保对产品质量的不良影响降到最低,变更实施前的研究结果是否可以充分证明变更前后产品质量的一致性或可比性等。
2.6清洁方面
口服固体制剂的生产过程常会涉及粉末物料,密封性是必须考虑的因素之一。根据所涉及的物料和工艺总体策略,物料处理系统将需要提供最低水平的密封。监管检查时可以关注该最低水平的确认验证是否全面考虑了所有组件和特性并应能避免污染和交叉污染,同时便于清洁 ;对操作人员潜在的职业暴露是否采取了预防措施,特别是在设备运行、配置和拆卸期间。
部分单元操作可能是非连续的,在物料处理设备或组件体积较小的情况下,可能会采用非在线清洗 (clean out of place,COP,即将设备拆卸清洗后重新组装 ) 的方式。COP 包括人工清洗或使用机器清洗,对这类清洗过程的监管检查可关注设备的可获得性、组装和操作的便利性,以及洗涤剂的选择、清洗液流速、清洗液对内表面的覆盖、超声的使用、清洗周期、清洗时间、清洗温度等方面的过程控制设置是否合理。对于不容易拆卸的大型药品连续制造系统或组件,通常会采用在线清洗 (cleanin place,CIP) 的方式,以减少对设备的拆卸组装,并减少停机等待的时间。CIP 操作可能会采用自动化系统、喷淋系统和浸渍等清洗方式,为确保覆盖度和密封性,应对相关具体细节进行确认。除上述对于 COP 的检查要点外,对 CIP 的检查还可关注是否合理设计并确认喷嘴的性能及位置、是否充分验证清洗程序等。
3、总结与展望
药品连续制造的物料处理是一个科技密集的工业过程,特别是口服固体制剂的连续化干燥和固体或流体的处理,难度较高,需要考虑存储条件、存储容器类型和特性、机械助流、研磨和筛分、采样和监测、气体输送、单元操作之间的接口、清洁等多方面细节,还需要延伸考虑系统的耐用性、密封性、可切换性和灵活性等。此外,并非所有单元的操作均可集成,可供选择的试验和中试设备也比较有限。因此,在生产线的整体设计和确认验证方面,研究工作的深度和广度一般远超过了批量制造,并额外需要统计学、过程控制、建模等多方面的专业知识和方法。连续制造对生产企业和监管部门都会带来较大的挑战,需要充分做好知识储备、加强沟通交流,结合物料处理的特点对相应风险加以额外关注。
相比批量制造技术来说,连续制造的物料处理需要对材料属性有更深的理解。监管检查中可关注企业是否建立并扩展了相应物料的质量标准,以及是否有针对性地开展了供应商管理。偏差管理也是监管检查关注的重点方面,特别是可能产生的不合格物料、中间品、成品的追溯和处置,对产品质量影响的风险评估,以及放行策略的合理性。此外,在连续制造商业化实施过程中,伴随着对产品和工艺理解的加深,可能涉及控制算法和模型的调整以适应原材料的变化,检查中可以关注其涉及的上市后变更研究是否符合相应指南的要求、是否在药品生产质量管理规范 (GMP) 体系下规范开展变更管理以及是否及时按要求申报注册、备案或报告等。
推动连续制造在制药行业的应用,有利于提高口服固体制剂产品的质量和可及性,降低患者用药负担。药品连续制造的发展需要行业协会、设备供应商,以及在连续过程和自动化方面具有专业知识的科研院所共同参与,在工艺技术提升的同时,质量保证技术也应当同步提升,充分运用 PAT 工具、创新检验方法和数字化质量保证等手段 [16-19],确保持续生产出符合质量要求的产品,从而充分推动我国制药工业迈向现代化、提升竞争力。
参考文献
[1] ICH.Continuous manufacturing of drug substances anddrug products, Q13 [EB/OL].(2021-07-27)[2021-10-08].https://database.ich.org/sites/default/files/ICH_Q13_Step2_DraftGuideline_2021_0727.pdf.
[2] WAHLICH J.Review: continuous manufacturing of smallmolecule solid oral dosage forms [J].Pharmaceutics, 2021,13(8): 1311.
[3] 胡延臣.药品连续生产及全球监管趋势[J].中国新药杂志, 2020, 29(13): 1464-1468.
[4] FDA.Quality considerations for continuous manufacturing:guidance for industry [EB/OL].(2019-02-01)[2021-10-08].https://www.fda.gov/media/121314/download.
[5] PMDA.Points to consider regarding continuousmanufacturing [EB/OL].(2017-05-01)[2021-12-06].https://www.nihs.go.jp/drug/section3/AMED_CM_PtC.pdf.
[6] PMDA.PMDA views on applying continuous manufacturingto pharmaceutical products for industry [EB/OL].(2018-03-30)[2021-10-08].https://www.pmda.go.jp/files/000223712.pdf.
[7] PMDA.State of control in continuous pharmaceuticalmanufacturing [EB/OL].(2018-05-01)[2021-12-06].https://www.nihs.go.jp/drug/section3/AMED_CM_CONTROLST.pdf.
[8] EMA.Continuous manufacturing - EMA perspective andexperience [EB/OL].(2017-09-19)[2021-12-06].https://dc.engconfintl.org/cgi/viewcontent.cgi?article=1033&context=biomanufact_iii.
[9] 曹 萌, 丁力承, 胡延臣, 等.药品连续制造全球监管发展现状与思考[J].中国药事, 2022, 36(4): 364-376.
[10] USP.Policy considerations to help harness pharmaceuticalcontinuous manufacturing for a more resilient medicinesupply chain.[EB/OL].(2021-04-01)[2021-10-08].https://www.usp.org/sites/default/files/usp/document/supply-chain/usp-policy-considerations-for-pharmaceuticalcontinuous-manufacturing.pdf.
[11] ASTM.Standard guide for application of continuousprocessing in pharmaceutical industry [EB/OL].(2016-12-27)[2021-10-08].https://www.astm.org/e2968-14.html.
[12] ISPE.Good practice guide: continuous manufacturing oforal solid dosage forms [EB/OL].(2022-03-30)[2022-05-11].https://ispe.org/publications/guidance-documents/good-practice-guide-continuous-manufacturing-oral-soliddosage-forms.
[13] 刘 飞, 李海燕.粉末直接压片连续制造缓释片[J].临床医药文献电子杂志, 2018, 5(15): 196-197.
[14] 袁春平, 时 晔, 王 健, 等.口服固体制剂连续制造的研究进展[J].中国医药工业杂志, 2016, 47(11): 1457-1463.
[15] BHALODE P, TIAN H, GUPTA S, et al.Using residencetime distribution in pharmaceutical solid dose manufacturinga critical review [J].Int J Pharm, 2021, 610: 121248.
[16] 张 闯, 唐文燕.过程分析技术在制药工业中的应用及监管考量[J].中国医药工业杂志, 2021, 52(3): 407-413.
[17] 董正龙, 曹 萌.信息技术在生物制药工业中的应用[J].中国医药工业杂志, 2019, 50(11): 1262-1267.
[18] 朱 馨, 陈桂良, 曹 萌.药品生产的数字化质量保证探索与实践[J].中国医药工业杂志, 2022, 53(3): 395-398.
[19] 曹 萌, 葛渊源, 张景辰, 等.药物分析新技术在药品科学监管中的应用[J].中国药事, 2021, 35(6): 614-623.
本文作者曹萌、葛渊源、胡延臣、王亚敏、曹轶,上海药品审评核查中心、国家药品监督管理局药品审评中心、国家药品监督管理局食品药品审核查验中心,来源于中国医药工业杂志,仅供交流学习。