您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-12 09:10
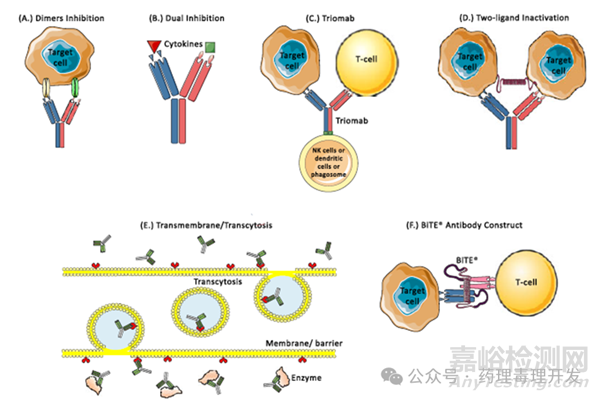
双抗又称为“双靶点”或“双特异性”抗体,可以结合同一细胞或不同细胞表面的不同靶点,也可以结合不同的可溶性靶点,甚至还可以是同一靶点的两个不同表位。常见的双抗作用示意图如下,图A代表同一细胞不同靶点,图B代表不同游离靶点,图C、D、F代表不同细胞的不同靶点,只不过Fc功能有所区别,图E则代表结合可内吞膜受体及内吞后的膜内靶点。
为实现双抗的多功能,其结构也是多种多样,如下表所示,有IgG样,也有非IgG类型的。

结构和功能的多样,为双抗的临床药理学研究也带来一些挑战。相较于传统的单抗,双抗的临床药理学方面也有些特殊考量。本文围绕双抗临床药理学研究展开讨论,并分享若干案例。
PK/PD
FDA在2021年颁布的《Bispecific Antibody Development Programs Guidance for Industry》中提到双抗的活性形式可能是个混合物,既包括有活性的组分(如未结合配体、结合1个配体的形式)和失活的组分(如不能结合配体的形式),应结合PD指标,鉴定出与PD最相关的组分。因此,可能需要开发不只一个方法以鉴定多种形式的分子。有时也可用PD方法代替,比如后续案例中的Blinatumomab就属于这类情况。
另外,双抗引起的免疫反应,有可能只是针对其中1个结构域,抑制了1个靶点的功能,另一个靶点的功能却是完整的。因此,有可能需要开发针对不同双抗结构域免疫反应的检测方法。考虑到双抗结构的多样性及复杂性,FDA鼓励企业就特定产品的开发计划与其临床药理部门进行沟通。
免疫原性、生物标记物
双抗作为治疗性蛋白,也会引发免疫反应。通常通过检测抗药抗体(antidrug antibodies, ADA)评估药物的免疫原性风险。但并不是所有抗体都会形成ADA,比如单抗rilotumumab的ADA发生率就是0%。双抗产品catumaxomab的ADA发生率也是0%,blinatumomab的ADA发生率为1%。影响双抗ADA形成的风险因素与其它单抗类似,包括双抗结构、是否存在外源序列、给药途经(SC较IV具有更高的ADA发生率)及患者免疫系统状态。ADA的形成会影响PK,继而影响PD。
双抗的生物标记物开发通常会面临诸多挑战。双抗需要兼顾两个靶点的生物学作用,甚至需要找到针对两个靶点的特定marker。比如BiTE抗体结构的blinatumomab,既需要证明T细胞的参与,又需要评价肿瘤靶点CD19的情况。前者可以通过检测急性淋巴细胞白血病患者中CD69和CD25表达的上调实现。
药物-药物相互作用(Drug-drug interaction, DDI)
有研究统计过FDA批准的68个治疗性蛋白新药,发现细胞因子调节是单抗类药物出现DDI的主要风险。细胞因子会影响肝脏CYP酶和药物转运体的表达。很多生物药物通过调节细胞因子水平,间接影响CYP酶和转运体。而细胞因子对CYP酶和转运体的影响程度,取决于细胞因子水平、细胞因子类型及细胞因子升高的持续时间。双抗药物很多是依赖T细胞发挥作用,典型的是以CD3-TAA为代表的分子。这类药物的免疫调节作用强,多数会出现细胞因子风暴,潜在的DDI风险较其它产品也高。
临床DDI研究可以通过群体药代动力学(Population PK, PPK)实现。可以将PPK嵌入Ⅱ、Ⅲ期大型临床试验中,从多个研究中汇集数据。
监管机构对于治疗性蛋白的DDI研究也给出过建议。2007年,EMA出台《Guideline on the Clinical Investigation of the Pharmacokinetics of Therapeutic Proteins》,指出免疫调节剂如细胞因子具备潜在诱导或抑制CYP酶的潜力,可以改变共同给药的小分子化药的代谢行为。建议对这类蛋白产品开展潜在的体内或体外研究,以确认该风险。
基于模型的临床药理学研究
通过对临床前PK参数数学建模预测临床PK在单抗中已经比较常见,常用的是异速缩放模型。该模型在线性PK的抗体中表现最好。当然,适用于非线性PK的模型也已经开发出来了。单抗的模型使用经验同样适用于双抗。比如EGFR-HER3双抗MEHD7945A就是采用食蟹猴二室模型获得PK参数,经定量药理模型(Species-invariant time method)转化后预测人体PK。
双抗的生物分析之前写过一篇单独的文章进行描述,有兴趣可翻看。双抗PK检测的独特性及已上市双抗PK检测方法汇总
分享几个案例。
案例一:Catumaxomab
Catumaxomab是一款CD3-EpCAM双抗,2009年被EMA批准上市,通过腹膜内给药途径,治疗肿瘤相关的恶性腹水。严格意义上讲,Catumaxomab应该算是一个三抗。如下图所示,Catumaxomab是由结合CD3的抗体、结合肿瘤抗原EpCAM的抗体以及结合Fc受体的恒定区组成,是具备Fc介导的ADCC、ADCP等效应功能的多抗。相较于EpCAM单抗,Catumaxomab的活性强1000多倍。
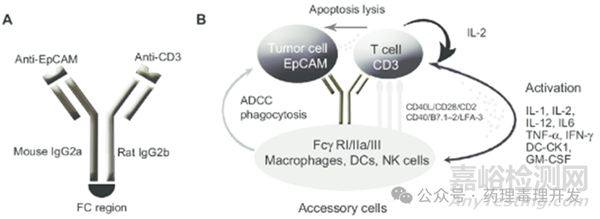
Catumaxomab首次人体临床试验采用的静脉注射给药,低剂量即出现了急性肝衰竭、细胞因子风暴相关的严重系统毒性。之后开发了腹腔注射给药途径,解决了因静脉给药大量非特异性T细胞激活导致的严重毒性反应,并显示了不错的药效。
Catumaxomab的生物分析采用ELISA方法,双抗夹心原理,包被的抗体大鼠IgGλ轻链特异性抗体,检测抗体为生物素标记的抗小鼠IgG2a抗体。此方法检测的总的药物浓度,包括所有靶点结合、非结合的药物形式。
PK/PD研究方面,恶性腹水患者接受4个剂量(10、20、50、150μg/kg)IP注射给药。腹水中可检测到高浓度的catumaxomab暴露。药物浓度随着给药剂量升高而增加,峰浓度可在末次药后19h观测到。消除半衰期约2.13天。PD反应通过检测细胞因子释放(TNF-α、IFN-γ、IL-2、IL-6、IL-10)得以确认。临床Ⅱ/Ⅲ期研究发现,相对淋巴细胞计数升高13%可以作为潜在良好预后的PD biomarker。此外,PD方面还观察到catumaxomab可以完成效应T细胞从外周血到组织的再分布,扩增EpCAM特异性T细胞,还能产生针对不同肿瘤抗原的杀伤活性。
由于catumaxomab的Fc采用的是大鼠和小鼠IgG,因此ADA风险潜在比较高。不过有意思的是,catumaxomab仅在末次药后检测到ADA,末次药前时间点并未发现ADA存在。另外,抗小鼠抗体阳性的患者的无穿刺生存期是要长于抗体阴性患者的。出现这一反常药效的原因,可能跟免疫原性更强的患者潜在具备更强的免疫反应有关。
案例二:Blinatumomab
如下图所示,Blinatumomab是BiTE双特异抗体结构,由CD3抗体和CD19抗体组成。作用机制也比较容易理解,就是利用患者自身的T细胞,通过CD3抗体再定位,杀伤自身CD19阳性的肿瘤细胞。Blinatumomab在pmol浓度即可激活T细胞,而且不依赖组织相容性复合体的抗原递呈和T细胞受体。
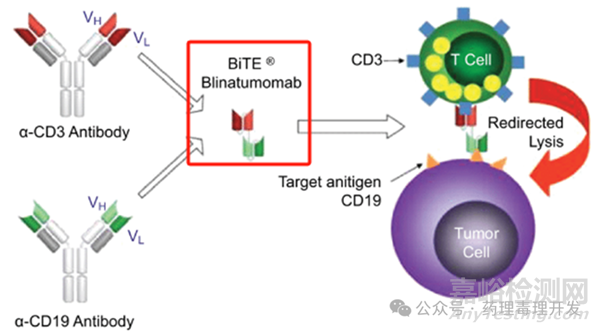
Blinatumomab与小鼠、大鼠和犬的抗原均不结合。为开展非临床研究,构建了抗鼠CD3和CD19的替代分子。虽然替代分子可以诱导小鼠T细胞激活,并裂解小鼠B细胞,能使细胞因子释放增加,与blinatumomab的作用机制类似。但是,Amgen并未基于替代分子的数据,采用传统方法计算FIH起始剂量,而是采用了“trial and error”的路径。个人理解的是“试错”。实际情况是,Amgen先开展了3个小规模的临床Ⅰ期预试验。Blinatumomab每周给药1次、2次、3次,每次输注2或4h。结果由于未见明显临床获益,且出现中枢神经系统和细胞因子风暴相关的不良反应(Adverse effect, AE),这三个试验均被终止。当然,也不是一无所获,研究中发现的细胞因子升高、轻度的T细胞激活和外周血B细胞的降低提示了blinatumomab的MOA是发挥作用的。此外,研究中出现的AE主要发生在给药开始阶段。该项目如果想继续推进,面临的难题是如何实现药效和毒性的平衡?Amgen团队认为blinatumomab的半衰期短,需要持续保持一定的有效暴露才能确保药效的发挥。另外,为解决AE问题,可以通过step dosing实现,同时给予糖皮质激素类药物预处理。
在后续扩展的Ⅰ期临床研究中,给药方案调整为0.5-90μg/m2/天,连续输注4或8周。具体方案分为两种,一种是0.5、1.5、5、15、30、60、90μg/m2/天flat dosing;一种是5-15、5-30、5-60、15-60μg/m2/天 1-step dosing、5-15-60μg/m2/天 2-step dosing,即先用低剂量诱导,个体内逐步提升剂量。结果显示step dosing更优,解决了前期面临的药效-毒性平衡的问题。
Blinatumomab的生物分析方法比较特别,采用的流式细胞术(FACS)检测的T细胞表面的CD69表达。CD69是T细胞活化的biomarker之一,且具有浓度依赖性。Amgen认为,基于细胞活性的方法相较于ELISA检测血药浓度是有优势的。个人比较好奇的是,细胞活性方法毕竟是间接反映blinatumomab暴露情况,能替代直接的血药浓度检测吗?CD69更像是PD指标,两者同时进行是否更好一些,既检测直接检测血药浓度的方法,同时观测CD69表达情况。
PK/PD方面,blinatumomab没有Fc,不涉及FcRn介导的再循环,进入体内后会被快速代谢成小肽,从外周快速清除。Blinatumomab在人体内呈线性PK,未见靶介导的消除。Blinatumomab主要在外周血中分布,表观分布容积3-5L,与血容量相当。Blinatumomab的消除半衰期约2h。PD方面,blinatumomab唯一激活的免疫细胞是T细胞。人体给予blinatumomab后,T细胞会出现增殖,之后分泌颗粒酶、穿孔素,杀掉肿瘤靶细胞。其它PD作用还包括,T细胞的再分布,剂量依赖性B细胞耗竭及一过性细胞因子升高。
体内ADA的产生需要B细胞的参与。Blinatumomab对B细胞的耗竭作用降低了其ADA风险。Blinatumomab的ADA阳性率仅为1%左右。个别患者能看到ADA对PK的影响,主要体现在血药浓度的降低。但是,由于ADA发生率太低,无法就ADA是否对安全性或有效性造成影响得出确切结论。
体外人体肝细胞实验显示blinatumomab不影响CYP450酶的活性。不过细胞因子对CYP450酶的表达有潜在影响,而blinatumomab会引起细胞因子一过性升高。体外研究显示,细胞因子鸡尾酒(多种因子混合)呈浓度和时间依赖性的抑制CYP3A4、CYP1A2、CYP2C9的活性。细胞因子抑制肝脏CYP450酶的主要因子是IL-6。采用PBPK模型预测IL-6对肝脏CYP450酶的抑制程度和持续时间。结果显示,临床峰浓度的IL-6可以抑制CYP3A4、CYP1A2、CYP2C9的活性达30%,持续一周左右。虽然DDI风险不高,但确实有可能造成CYP3A4、CYP1A2、CYP2C9为底物的药物暴露量增加。
案例三:Solitomab(AMG110, MT110)
如下图所示,Solitomab结构与blinatumomab类似,也是BiTE结构,由EpCAM和CD3单链抗体组合而成。
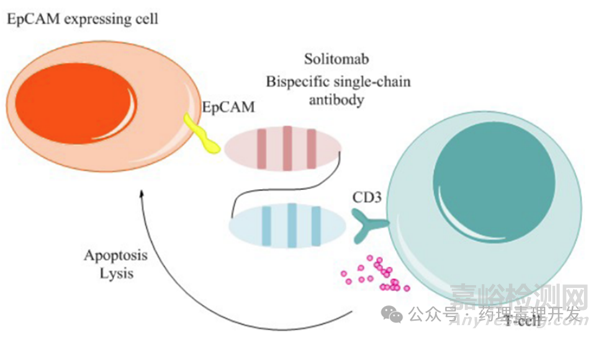
Solitomab的FIH剂量为1-96μg/天。1μg/天的起始剂量是采用替代分子在小鼠中的数据基于最低预期生物效应水平(minimal anticipated biological effect level, MABEL)获得。剂量爬升过程中出现的主要剂量限制性毒性是肝功能异常和严重的腹泻。为控制肝酶升高,采用的方案是从3μg/天开始,分步进行个体内剂量爬升,有点类似blinatumomab用过的策略。此外,给药前3天还需要给予糖皮质激素处理。严重腹泻限制了给药周期,使相对高剂量下的最长输注时间不能超过3或4周。
Solitomab的PK行为呈线性,24h内即达到稳态,消除半衰期约4.5h。96μg/天对应的Cmax约为6ng/mL。长期重复给药未引起T细胞失能或损害T细胞的效应功能。7/63(11%)患者检测ADA,其中2例患者发现PK行为的改变。ADA阳性的患者未见过敏或其它超敏反应症状。

来源:药理毒理开发


