您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-12 18:43
一、基本概况
1、自然环境
日本位于太平洋西岸,是一个由东北向西南延伸的弧形岛国。西隔东海、黄海、朝鲜海峡、日本海与中国、朝鲜、韩国、俄罗斯相望。陆地面积约37.8万平方公里,包括北海道、本州、四国、九州四个大岛和其它6800多个小岛屿。属温带海洋性季风气候,终年温和湿润。
2、人口和行政区划
日本人口约1亿2339万(2024年1月)。主要民族为大和族,北海道地区约有1.3万阿伊努族人。通用日语。主要宗教为神道教和佛教。日本65岁以上老龄人口数约占人口总数的三成,是世界上人口密度最大和老龄化最严重的国家之一。
日本全国划分为1都(东京都:Tokyo)、1道(北海道:Hokkaido)、2府(大阪府:Osaka、京都府:Kyoto)和43县(省),下设市、町、村。首都东京是日本的政治、经济、文化中心,人口约1411万(2024年1月)。
3、2024年出口概况
日本是世界经济强国。据统计,2023年日本实际国内生产总值(GDP)约559万亿日元,同比增长1.9%。2024年1-6月,中国向日本出口医疗器械总计约111.74亿元人民币,同比下降5.34%。
二、日本医疗器械监管机构和法规要求
日本医疗器械的监管机构为厚生劳动省(Ministry of Health, Labor and Welfare, MHLW),其与医疗器械监管有关的主要职责有:①制定医疗器械监管政策,②注册批准的最终审查,③产品退市。
日本独立行政法人医药品与医疗器械综合机构局(Pharmaceuticals and Medical Devices Agency, PMDA)是一个独立的行政机构,其与厚生劳动省合作,以确保医疗器械的安全和质量;PMDA协助MHLW医疗器械监管政策实施的职责包括:1)医疗器械审批审核,2)QMS/GLP/GCP检查,3)不良事件报告的收集和分析。
日本医疗器械注册需要遵循《药品与医疗器械法》(Pharmaceutical and Medical Device Act,缩写为PMD Act)。
三、医疗器械定义
根据PMD Act第2条第4项,医疗器械定义为:用于诊断、治疗或预防人类或动物疾病或影响人类或动物身体结构或功能的试剂、耗材和设备。
“この法律で「医療機器」とは、人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は人若しくは動物の身体の構造若しくは機能に影響を及ぼすことが目的とされている機械器具等(再生医療等製品を除く。)であつて、政令で定めるものをいう。”
根据PMD Act第2条第14项,体外诊断试剂定义为:专门用于疾病诊断的药物,且该药物不直接用于人体或动物体内。
“この法律で「体外診断用医薬品」とは、専ら疾病の診断に使用されることが目的とされている医薬品のうち、人又は動物の身体に直接使用されることのないものをいう。”
四、产品分类与符合性路径
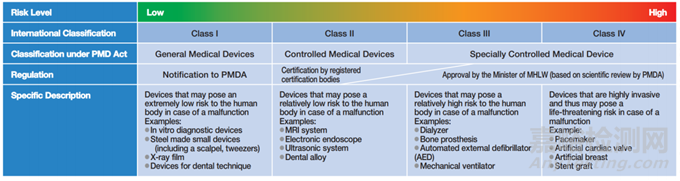
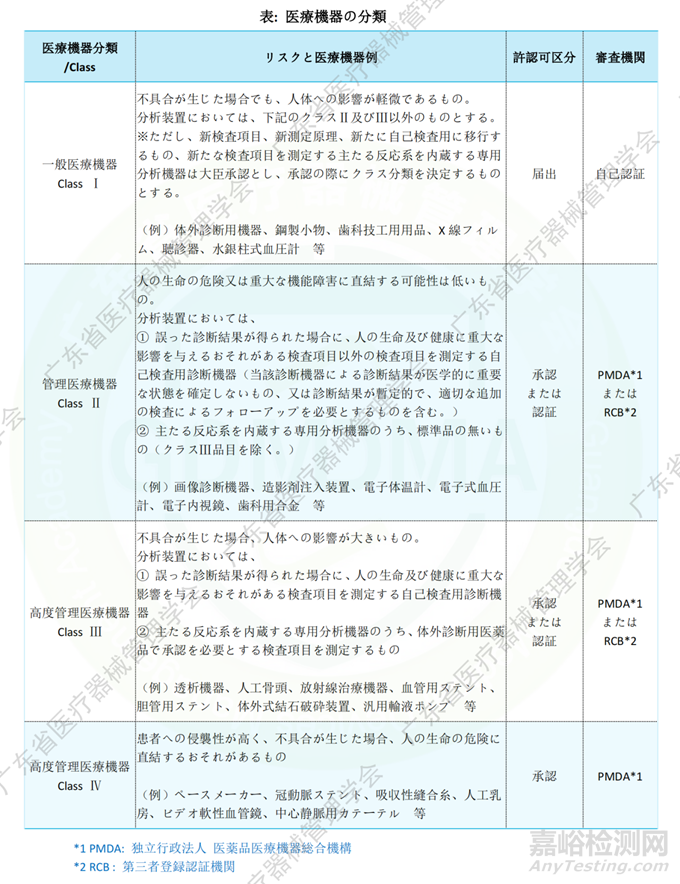
医疗器械根据其对人类造成的危险程度进行分类,分为一般医疗器械(Class I)、管理医疗器械(Class II)和高度管理医疗器械(Class III, IV)。医疗器械分类不同,其符合性路径不同;即Class I仅需向PMDA备案(届出),大部分Class II及小部分Class III(有产品标准)需提交J-RCB(日本第三方公告机构)认证,而小部分Class II、大部分Class III及所有Class IV需经PMDA审核后MHLW承认。
五、注册流程
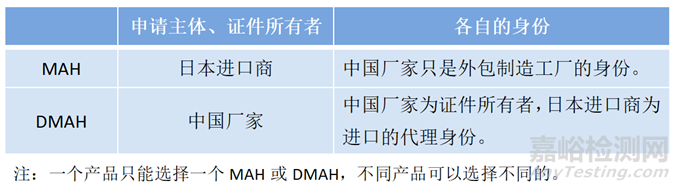
1、注册主体
2、注册流程

备注:
1)符合资质的J-RCB名单可检索登録認証機関;
2)产品相关证书无有效期,工厂注册证书及QMS证书有效期均为5年;
3)QMS符合性审查申请可于产品注册申请受理后10个工作日内提交。
3、注册资料
1)工厂注册:企业名称及地址,企业法定代表人简历(如提供原文及日文翻译件,且如原文不是英文,需同时提供翻译者资质证明),生产场地平面图。
2)产品备案:提交“医療機器製造販売届書”,即备案申请表(日文)。
3)产品认证需提交资料目录清单视具体的J-RCB要求,符合资质的J-RCB名单可检索登録認証機関。
4)认证基准
认证基准需要日本官网查询, 类似国内的指导原则, 需要确定产品需要满足哪些 JIS 标准。PMDA有发布认可协调标准清单,一般来讲通用型的标准都认为IEC和ISO的标准,性能标准部分有发布不少JIS标准,特别是没有国际标准的产品。
5)产品承认


备注:申请表及技术文件摘要(STED)须以日文提供,测试报告、临床试验报告等附件资料可以英文提供。
6)QMS符合性审查 :以下为PMDA要求,至于J-RCB审核情况需视具体的J-RCB要求,符合资质的J-RCB名单可检索登録認証機関。日本的质量体系法规为MHLW Ministerial Ordinance No. 169, 2004及修订案2021(对于外国制造商来说,基本上等同于ISO 13485的要求)。
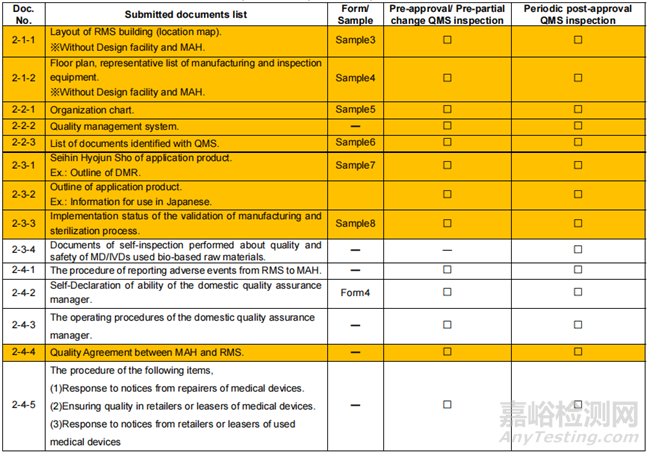
◆ 申请提交资料清单
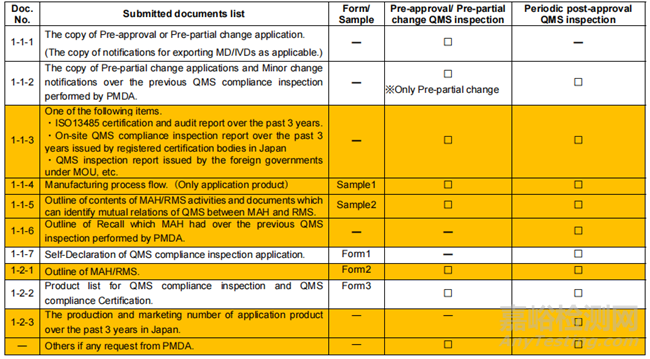
※Documents highlighted in orange is related to RMS.
※MAH: Marketing Authorization Holder. RMS:Registered Manufacturing Site.
◆ 线上审核资料清单
通常,审查机构根据提交的QMS符合性审查申请资料决定采取线上还是现场审核方式。
※ Documents highlighted in orange is related to RMS.
※ MAH: Marketing Authorization Holder. RMS:Registered Manufacturing Site.
※ MDSAP可豁免现场审核,及简化线上审核;相关信息可检索MDSAP報告書の利用申請について。
◆ 现场审核安排
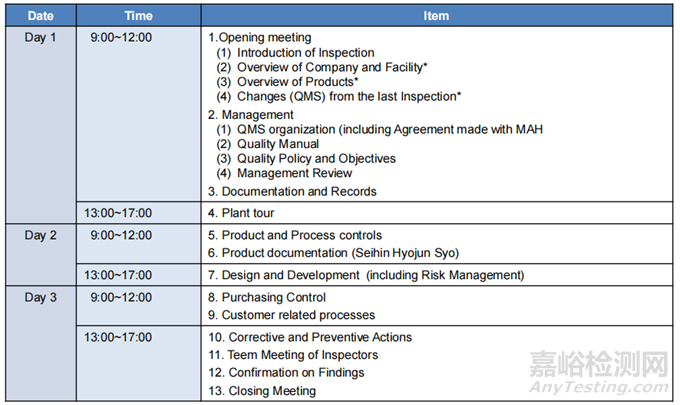
Note: As to items with*, Please give presentations to Inspectors.
4、注册周期及费用
工厂注册:1个月;90,000円
产品备案:即时,免费
产品认证:视具体的J-RCB情况,符合资质的J-RCB名单可检索登録認証機関
产品承认:4∼12个月,费用可见手数料
QMS符合性审查:约6个月,费用可见QMS適合性調査手数料計算
5、注册提交
PMDA及MHLW注册申请有关业务的提交方式可选择FD申請或DWAP申請,感兴趣的同行可检索:
FD申請、DWAP申請
备注:各类别医疗器械制造贩卖许可证书的有效期如下表:

6、该区域有关UDI的要求
日本已于2022年12月全面强制实施UDI要求,包装赋码要求如下:
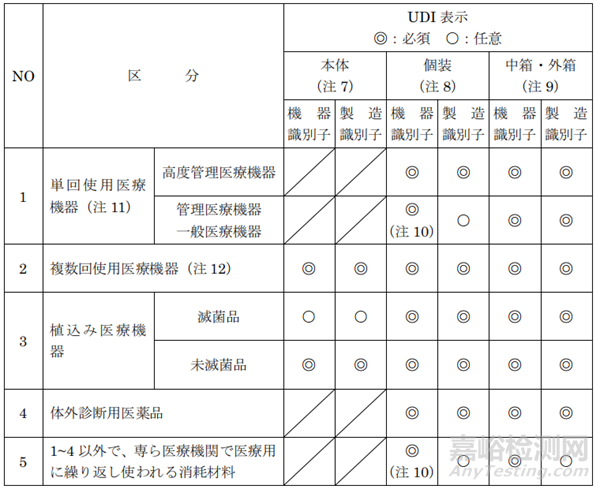
UDI申请及相关赋码格式等要求详情,可检索GS1 Japan。
通常,格式选用GS1-128条形码,对于体积较小且又需在本体上直接打码的器械可选用GS1二维码。

医疗器械获得市场准入后出货前,MAH或DMAH需将UDI码上传“医療機器データベース”,该数据库面向企业的常规操作功能免费,账号创建网址链接。
7、其他注意事项或特别提醒
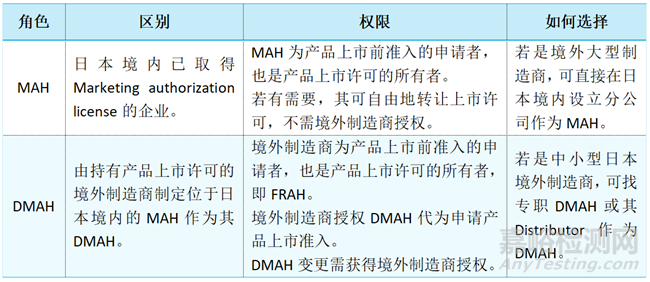
1)MAH资质
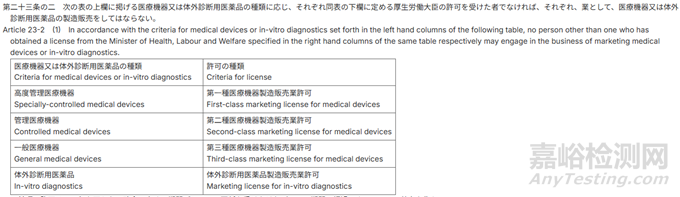
2)DMAH与MAH差异


来源:广东医疗器械学会


