您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-01-10 20:12
一、基本概况
1、自然环境
韩国位于亚洲大陆东北部朝鲜半岛南半部。东、南、西三面环海,国土面积为10.329万平方公里。属温带季风气候,年均气温13℃-14℃,年均降水量约1300毫米-1500毫米。
2、人口和行政区划
韩国人口约为5100万。为单一民族,通用韩国语,50%左右的人口信奉佛教、基督教、天主教等宗教。
全国划分为1个特别市:首尔特别市;2个特别自治市(道):世宗特别自治市、济州特别自治道;8个道:京畿道、江原道、忠清北道、忠清南道、全罗北道、全罗南道、庆尚北道、庆尚南道;6个广域市:釜山、大邱、仁川、光州、大田、蔚山。首都为首尔(Seoul),人口约938万。
3、2024年出口概况
2024年1-10月,中国向韩国出口医疗器械总计约90.77亿元人民币,同比上升4.20%。
数据来源:广州众成大数据科技有限公司
二、医疗器械监管机构和法规要求
韩国食品药品安全部(Ministry of Food and Drug Safety,MFDS)负责韩国的医疗器械监管,为减少监管工作压力,MFDS下放了部分监管权力或与有资质第三方审查机构合作;如将I类医疗器械告知以及II 类认证的受理/审批工作授权了其附属机构,即国家医疗器械安全信息中心(National Institute of Medical Device Safety Information,NIDS);此外,就II 类认证的技术文件审查工作及KGMP符合性审核工作与有资质的第三方审查机构(TPR)进行合作。
韩国医疗器械的监管法规体系可分为以下四个层级:

韩国医疗器械注册需要遵循如下法规要求:
◆ ACT ON IN VITRO DIAGNOSTIC MEDICAL DEVICES,体外诊断器械法案
◆ DECREE OF THE ACT ON IN VITRO DIAGNOSTIC MEDICAL DEVICES, 体外诊断器械法案执行令
◆ ENFORCEMENT RULE OF THE ACT ON IN VITRO DIAGNOSTIC MEDICAL DEVICES,体外诊断器械法案实施条例
◆ Regulation on the Permission, Notification, Review, Etc. of In Vitro Medical Devices,体外诊断器械许可、认证及告知审查规范
◆ ACT ON MEDICAL DEVICES,医疗器械法案
◆ DECREE OF THE ACT ON MEDICAL DEVICES,医疗器械法案执行令
◆ ENFORCEMENT RULE OF THE MEDICAL DEVICES ACT,医疗器械法案实施条例
◆ Regulation on the Permission, Notification, Review, Etc. of Medical Devices,医疗器械许可、认证及告知审查规范
◆ In Vitro Diagnostic Medical Device Manufacturing and Quality Control Standards,体外诊断器械质量体系标准规范(KGMP)
◆ Medical Device Manufacturing and Quality Control Standards,医疗器械质量体系标准规范(KGMP)
三、医疗器械定义
根据“医疗器械法案”第2条,医疗器械定义为:供人类或动物单独或组合使用的仪器、机器、器械、材料、软件或任何其他类似产品,但《药事法》规定的药品和类似药品以及《残疾人福利法》第65条规定的残疾人辅助器具中的假肢和辅助器具除外:
a. 用于诊断、治愈、缓解、治疗或预防疾病的产品;
b. 用于诊断、治愈、减轻或纠正伤害或损伤的产品;
c. 用于测试、替换或转换结构或功能的产品;
d. 用于控制受孕的产品。
根据“体外诊断器械法案”第2条,体外诊断器械定义为:对人体或动物样本进行体外试验而单独或与组合使用的下列医疗器械,如试剂、造影剂、校准品、设备、仪器、器具及软件:
a. 用于生理或病理诊断的产品;
b. 用于确定疾病原因或观察疾病预后的产品;
c. 用于提供出生缺陷信息的产品;
d. 在组织移植或献血的情况下,用于提供判断安全性和相容性性所需信息的产品;
e. 用于预测治疗反应和治疗结果的产品;
f. 用于确定治疗方法或监测治疗效果或副作用的产品。
四、产品分类
根据“医疗器械法案实施条例”第2条,医疗器械按风险高低可分为Ⅰ类∼Ⅳ类。

根据“体外诊断器械法案实施条例”第2条,体外诊断器械按风险高低可分为Ⅰ类∼Ⅳ类。

五、注册流程
1、注册流程图

II*:符合以下情况的II类设备必须由MFDS审核:(1)需要临床实验报告;(2)数字医疗相关(如远程医疗系统);(3)命名或分类规则未定义的新设备;(4)与药品结合使用。
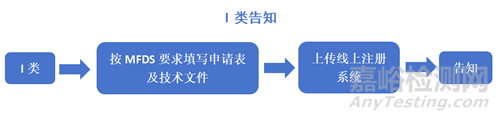
产品认证或许可流程:
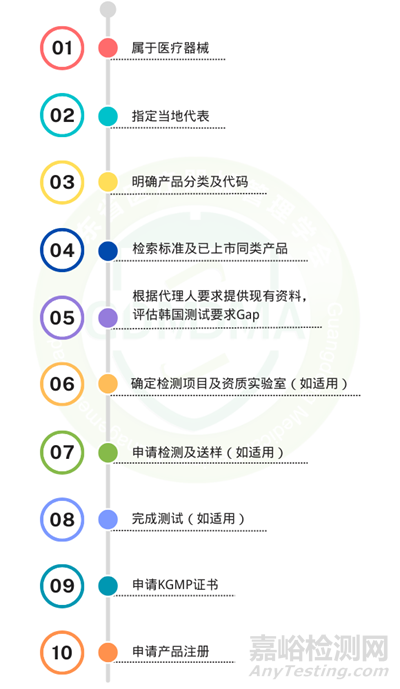
注:KGMP证书申请及产品注册申请可以同时进行,仅需在注册申请最终审批前拿到KGMP证书即可。
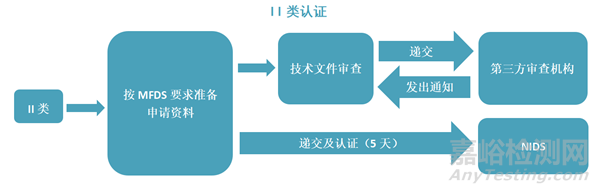

注:产品认证或许可证书有效期为5年,到期前6个月申请延续。

注:I类仅需符合KGMP条款4.1, 4.2, 5.5, 6.4, 7.1, 7.4, 7.5,7.6, 8.2.1, 8.2.2, 8.2.3, 8.2.6, 8.3, 8.5,其KGMP证书豁免定期延续审核,但仍需进行变更或附加审核;II、III、IV类KGMP证书有效期为3年,到期前90天申请延续。
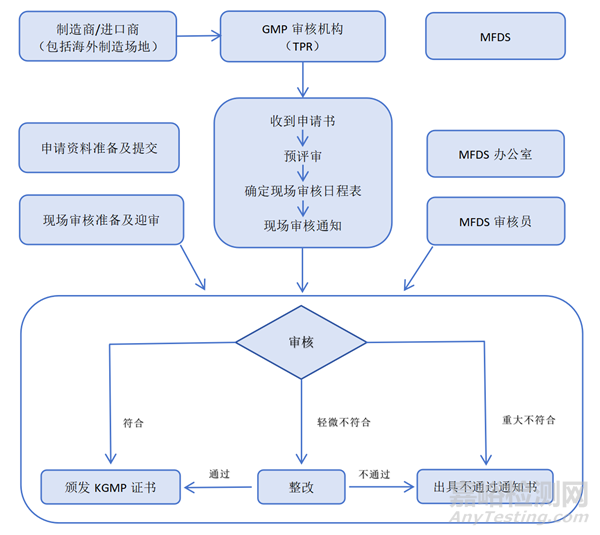
GMP Audited Bodies(TPR)
2、注册提交文档



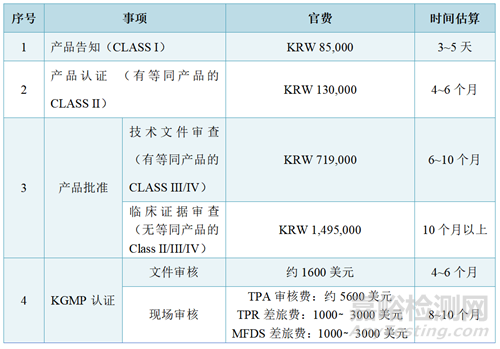
3、注册官费与周期估算
4、注册提交方式
通过韩国e-Government门户进行告知或申请递交。
5、现场审核特别提醒
KGMP从2023年新冠疫情后开始恢复现场审核,如无特殊情况(如严重火灾、洪水、地震或其他行政限制因素等),大多数外国制造商均需进行现场审核准备;但仍有可能通过MDSAP豁免首次KGMP证书申请的现场审核,除非制造商存在重大缺陷或安全问题、生产多款医疗产品或含人体组织/衍生物器械;KGMP证书失效期同MDSAP证书失效期。
6、该区域有关UDI的要求
韩国已全面实施UDI要求,包括Ⅰ类∼Ⅳ类;上市流通前,制造商或进口商需将UDI-DI以及与之关联的基本追溯信息上传至MFDS的UDI数据库Integrated Information System (IMDIS),上传信息具体要求可参考“Regulations on Unique Device Identification Management, Etc.”,如上传信息有变更,需于变更后10天内修改数据库相应信息。需特别提醒的是,MFDS要求同时上传医保信息(如是否在医保目录及医保代码等)。
MFDS认可GS1标准、HIBCC标准、ICBBAA标准此三种国际标准;其中,ICBBAA标准是用于全球血液制品的一种标准,HIBC标准原为美国的医疗产品专用编码标准(HIBC标准在中国没有代理机构);因此,除了血液制品管理外,中国厂商出海为尽量简化UDI实施工作,建议采用GS1标准。
出口韩国医疗器械Ⅰ类赋码到DI即可,其他类别需赋码到DI+PI;需特别提醒的是,为避免歧义,PI赋码格式统一要求为YYMMDD方式;其他要求可参照GS1标准。
7、韩国当地代表
当地代表可以是韩国当地子公司、进口商、经销商或第三方独立持证人(Korean License Holder, KLH)。
7.1 任职要求
1) KLH必须任命一名质量经理来监督其质量标准及法规的遵守情况;
2) KLH必须在MFDS上正式注册;
3) 必须持有有效的医疗器械进口经营许可证;
4) 必须是韩国公民或韩国注册企业。
7.2 职责
1) 递交医疗器械产品注册登记申请;
2) 递交韩国生产质量管理规范(KGMP)证书申请,并在现场提供协助;
3) 与经销商合作,共同获取同一产品注册登记证明,进口已注册登记产品;
4) 协调处理韩国事故报告事宜;
5) 按韩国食品药品安全部(MFDS)要求,作为首要联络人;
6) 如需要,协助提交保险费用报销(HIRA)申请,等。
7.3 同一产品注册(IPR)
由于韩国法规是当地代表持证与进口商角色合并,故为避免其可能对进口业务扩展造成潜在的牵制风险,制造商如在当地无子公司,一般宜选择第三方独立KLH +一个或多个进口商IPR方式,以确保上市前后法规符合性及销售业务的相对可控性。同一产品注册(IPR)不适用于Ⅰ类告知,可单独进行KGMP,仅注册技术文件需KLH配合,注册时间更短、费用更低。

来源:广东省医疗器械管理学会


