您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-05-26 06:05
药品三大属性,安全、有效、质量可控;不同开发阶段要求程度不同,总体方向为~安全性证据越来越多、有效性数据越来越充分、质量控制越来越严。那么,早期的创新药,尤其是IND阶段的品种,药学质量控制如何支撑其药物属性特点和政策法规要求?笔者根据自身的项目开发经验,及法规指南的解读,总结此文,以期共同进步。
01 创新药IND阶段药物特点
一个药物的发现,从最早的Hit、到逐渐分类的Lead、再到开始砸钱的PCC,经过一系列的分子生物学、药物化学、药理学、药物分析、药剂学、制药工程等众多学科的投入,以及数千万至亿RMB的经济成本投入,幸运的分子,将进入到一个品种的重要时间节点~IND。
这一过程,哪个学科重要?哪个学科地位高?大部分的争议,会在分子生物学、药理和药物化学之间进行讨论,功劳簿都会在“0到1”、“设计发现”、“源头基础”进行争论。对此,笔者不予发表观点,而是重点说一下从始至终的一个“眼睛学科”,保驾护航的学科,甚至是吃力不讨好的学科~质量。并着重讨论下,早期的质量研究,要将品种的质量控制到什么程度。

图1.1 创新药生命周期特点
02 创新药IND阶段政策&法规&指南
自从国家鼓励新药创制以来,无论是政策、法规、还是指南,其更新和迭代速度大大提升,为创新药的研发提供了大量的理论支撑。如IND阶段相关的《新药I期临床试验申请技术指南》、《化学药品创新药I期临床试验申请药学共性问题相关技术要求》、《化学药品I期临床试验申请药学研究信息汇总表》,等等。
➣ 新药I期临床试验申请技术指南
为帮助新药注册申请人申请I期临床试验,提高新药研发与审评效率,保护受试者安全与权益,保证临床试验质量,发布该指南。指南阐述了新药在我国开展首次临床试验时需要向CDE提供的信息。指南的目的是:明确新药I期临床试验的技术要求,提高I期临床试验申报资料的质量;通过规范I期临床试验资料的数据要求,缩短新药研发周期,加快新药上市进程。

图2.1 CDE关于《新药I期临床试验申请技术指南》的发布
➣ 化学药品创新药I期临床试验申请药学共性问题相关技术要求
对于I期临床试验申请,为了保障受试者的安全,药学审评通常重点关注与安全性相关的问题,例如杂质、稳定性、无菌制剂生产条件和除菌/灭菌方法、以及临床前动物安全性评价试验与后续人体临床试验所用样品的质量可比性等。上述的《新药I期临床试验申请技术指南》对相关药学研究内容和资料提交要求已经进行了阐述,但是审评中发现部分创新药I期临床试验申请仍然存在一些与上述安全性内容相关的药学问题。为了更好地实施国家局50号公告,促进创新药的研究和开发,本技术要求对创新药I期临床试验申请药学共性问题进行总结,以供申请人参考。

图2.2 CDE发布《化学药品创新药I期临床试验申请药学共性问题相关技术要求》
➣ 化学药品I期临床试验申请药学研究信息汇总表
《化学药品I期临床试验申请药学研究信息汇总表》,除了基本信息外,重点对原料药和制剂的药学信息进行抽取。原料药的质量控制,需要对关键理化特性、质量控制(对项目和限度进行汇总)、批分析、稳定性进行信息汇总。制剂的质量控制,需要对质量控制、批分析、稳定性进行汇总。
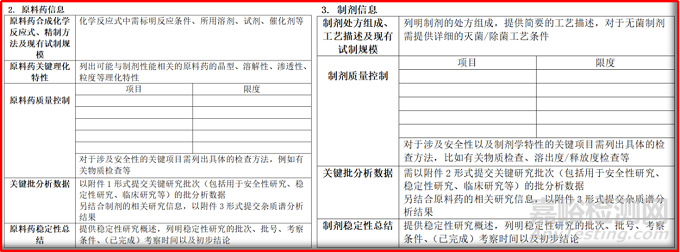
图2.3 《化学药品I期临床试验申请药学研究信息汇总表》摘取内容
除上述的原料药和制剂的2个重点表格外,还需要对原料药批分析数据(包括试制时间、地点、规模、工艺、用途、关键质量数据)、制剂批分析数据(包括试制时间、地点、规模、处方、工艺关键质量数据)、杂质谱分析,进行汇总。这里强调的是,要对杂质谱分析进行细致的联动研究,尤其是原料药-制剂-非临床之间的数据和试验关联。
03 创新药IND阶段具体质量控制
通过对上述新药I期临床IND的相关背景介绍,下面将从药学的角度,来具体分析讨论创新药I期临床IND申请,相关的药学质量研究及控制。
➣ 新药I期临床试验申请技术指南
对于新药申请Ⅰ期临床研究的药学研究资料,应遵循药物研发的规律,重点关注对计划研究的受试者安全性相关的药学研究信息(如基于现有知识对杂质谱的解析,有关物质检查专属性、灵敏度的方法学验证,潜在遗传毒性杂质分析和控制,生物新药的免疫原性和免疫毒性等)。也就是说,IND阶段质量控制的主要目标,就是一切与可能存在安全性隐患相关的所有因素,包括原料药和制剂。
表3.1.1 化药CMC相关的质量控制和稳定性研究

以上(包括全文),主要是针对化学药物,为了更好的理解质量在创新药生命周期中的作用,这里再略介绍下生物制品I期临床试验申请需要的质量控制。
对于生物制品,首先在结构确证方面即存在与小分子的很多不同之处。研发早期,应对样品进行初步结构确证,提交研究数据。完整的结构确证数据可在申报新药上市时提交,包括一级结构、二级结构和高级结构等。应提供结构确证用样品的批号,如用到对照品,应说明对照品来源、批号、数量。此外,对于涉及生物毒性、免疫原性的杂质,应提供敏感、特异的检测方法,拟定严格的控制要求。
表3.1.2 生物药相关的质量控制和稳定性研究

➣ 化学药品创新药I期临床试验申请药学共性问题
这些年,随着每年新报IND品种数量由数十、到一百、二百的增加,问题自然积累的越来越多。不同申办方&申请人,对不同领域的创新品种,理解不同、开发深度不同。对此,药审中心发布了《化学药品创新药I期临床试验申请药学共性问题相关技术要求》。
对于质量控制,重点对“质量标准&分析方法”、“杂质研究&控制”、“致突变杂质研究”、“稳定性试验”进行了要求,详细要求见下表。
表3.2 创新药I期临床试验申请药学共性问题

04 阶段小结&小悟
综上,对创新药IND阶段药学研究质量控制,进行了框架式的介绍,初步可了解该阶段质量控制的一般性要求。也许有读者会问,是否可以明确质量控制具体项目的具体程度和限度,比如,分析方法是否有必要做全套?一般杂质是否必须控制在0.1%以下?基因毒杂质需要研究到何种程度?稳定性试验到底做几个月?根据笔者多个项目的研究经验,答案真的是“具体问题-具体分析-具体回答”。因为,任何一个子项目都需要联动具体的品种特点、安全性数据支持、临床开发计划,等等。至于开发到何种程度,其实也无外乎宽松和严格两种,这与申办方的研发投入和项目开发经验息息相关。而想真正的对大部分项目进行全时空的把控,最终还是要落到各个项目的历练当中;一定的理论学习,和所谓的QbD,都是需要在经验的基础上,才能升华;否则,都是空谈。

来源:新浪医药


