您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-06-06 21:26
欧盟 CE法规
|
名称 |
法规 |
|
医疗器械法规 |
2017/745,MDR |
|
体外诊断器械法规 |
2017/746,IVDR |
欧盟 CE标准
|
名称 |
协调标准 |
|
质量管理体系 |
EN ISO 13485 |
|
包装 |
EN 868-X |
|
生物学评估 |
EN ISO 10993-X |
|
灭菌 |
EN 550,552,554,556 |
|
临床调查 |
EN ISO 14155-X |
|
风险分析 |
EN ISO 14971 |
|
标签&符号 |
EN 1041 & ISO 15223 |
|
医用电气安全 |
EN 60601-1 |
注册流程
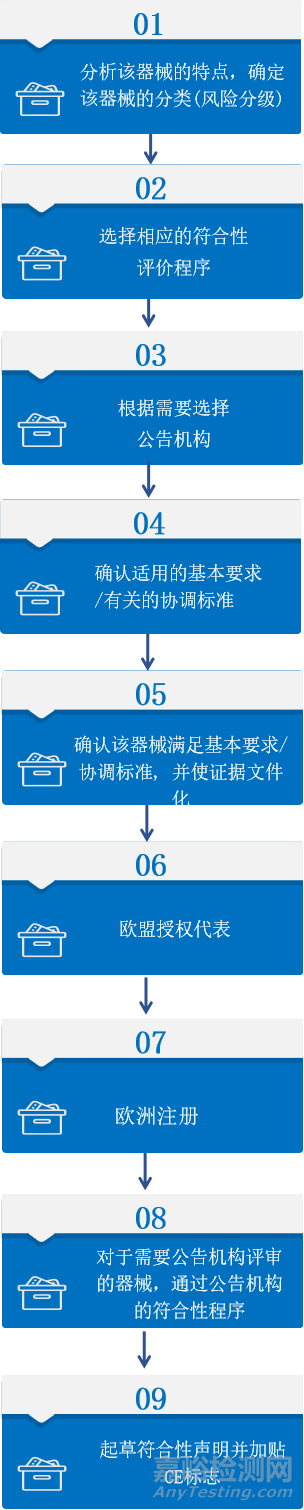
(A类非灭菌产品可直接通过自我声明途径)
技术文档
医疗器械CE注册中,技术文档的准备非常重要,根据医疗器械法规2017/745要求“技术文档”可能包含:企业简介及欧洲代表名称、联系方式;CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的证明材料),主要内容如下:
1.器械说明与性能指标:包含附件、前代和类似器械信息。
2.制造商提供的信息:
产品资料和控制文档;
产品生产工艺流程图;
产品的清洗消毒灭菌方法和确认的描述;
灭菌验证;
产品质量管控;
产品稳定性和有效期的描述。
3.设计与制造信息:
技术性能参数;
产品配合使用的附件、配合件和其它设备清单;
产品的图示与样品;
产品所用原材料及供应商;
包装材料说明;
包装验证;
标签;
使用说明书。
4.通用安全与性能要求:产品检验报告及相关文献,包含其符合附录I提供的通用安全与性能要求的证明资料。
5.风险利益分析和风险管理:产品潜在风险测试报告及相关文献
6.临床评价资料:
临床评价:通过临床文献资料、临床经验数据、临床试验等信息对产品是否满足使用要求或者适用范围进行的确认。
临床试验:临床试验资料包括临床试验方案和临床试验报告。
7.上市后监管计划
8.上市后监管报告或定期安全性更新报告(PSUR)
9.符合性声明文件
10.CE符合性标志
11.器械唯一识别标志(UDI)
12.欧盟授权代表

来源:中检华通威


