摘 要 Abstract
目的:对药品上市后安全性研究(PASS)政策执行情况进行分析,为优化PASS 政策提供参考。方法:基于史密斯模型,从理想化政策、执行机构、目标群体、政策环境等4 个影响因素出发,定性分析PASS 政策执行情况。结果:通过史密斯模型的4 个变量对PASS 政策执行影响因素进行分析,建议完善PASS 政策,出台PASS 管理规范和条例;加强对PASS 的监管,加大对PASS 的投入;加强药品上市后风险管理;健全药品上市后安全性评价体系等。结论:随着新政策的出台,我国对药品上市后安全监测愈加重视,加强PASS 有利于促进药品安全监管,更好保障公众健康及生命安全。
Objective: To analyze the implementation of post-authorisation safety study (PASS) policy and provide reference for optimizing the PASS policy. Methods: Based on the Smith model, the PASS policy implementation is analyzed qualitatively from four influencing factors: idealized policy, implementation agency, target group and policy environment. Results: The analysis of the factors influencing the PASS policy implementation through the four variables of the Smith model, introduce the management norms and regulations of PASS; strengthen the supervision of PASS, increase the investment in PASS; strengthen the risk management after marketing; and improve the safety evaluation system after marketing. Conclusion: With the introduction of the new policy, China pays more and more attention to the safety monitoring of drugs after the market. Strengthening PASS can promote the supervision of drugs, so as to ensure the life safety of patients and public health.
关键词 Key words
药品上市后安全性研究;政策执行;史密斯模型;药品不良反应监测;药物警戒
post-authorisation safety study; policy implementation; Smith model; adverse drug reaction monitoring; pharmacovigilance
药品上市后安全性研究(post-authorisation safety study,PASS)是指药品上市后,为保障用药安全对药品安全风险进行识别和评价风险控制措施效果的研究,是药物警戒中不可或缺的一环[1]。2007 年7 月,原国家食品药品监督管理局发布《药品注册管理办法》[2],提出要对上市后新药进行安全性管理。2011年5 月,原卫生部发布的《药品不良反应报告和监测管理办法》[3]规定,对于处于监测期的国产新药,应监测和报告此药上市后的不良反应,对于首次进口5 年内以及可能存在安全隐患的高风险药品,应开展重点监测,这在一定程度上推动了我国药物警戒的发展和突发不良反应事件预警制度的建立。2017 年6 月,原国家食品药品监督管理总局加入国际人用药品注册技术协调会(ICH),这对我国药品监管制度提出了更高层次的要求。2019 年8 月,新修订《药品管理法》首次明确提出建立药物警戒制度[4],我国药物警戒的发展前进了一大步。2020 年1 月,国家市场监管总局发布的《药品注册管理办法》[5] 规定,药品上市许可持有人(MAH)应当主动开展药品上市后研究,加强对已上市药品的持续管理;2021 年5 月,国家药品监督管理局(以下简称国家药监局)发布的《药物警戒质量管理规范》[6](GVP) 阐释了PASS 的定义,并对药品上市后安全性研究有关细节作出详细规定。为保障公众用药安全,我国药品相关法律法规不断完善,药品安全主体责任也更加明确。
本文基于史密斯模型,分析PASS 政策框架及执行过程中的影响因素,通过对4 个影响因素进行分析并提出相应建议,以期为PASS 政策执行的优化提供参考。
1、理论模型
1.1 史密斯政策执行过程的分析框架
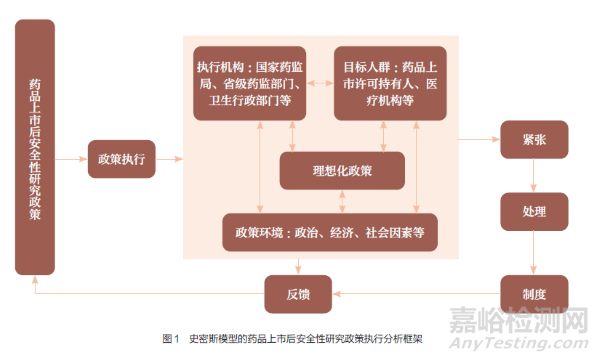
20 世纪70 年代,美国学者史密斯在《政策执行过程》一文中,首次提出分析政策执行过程及其影响因素的理论模型,该模型称为“史密斯模型”,又称“史密斯政策执行过程模型”。史密斯认为,政策执行受多方因素影响,主要体现在理想化政策、执行机构、目标群体、政策环境等4 个变量,这4 个变量紧密相关,彼此互相作用与影响。史密斯模型通过分析上述4 个变量之间的关系,使政策分析的脉络更加清晰,能用于更准确地分析和解决政策执行过程中的问题。首先,理想化政策是指在毫无干扰的环境下达到的完美状态,现实中难以实现,这是政策执行前最重要的一环。其次,执行机构是指政府中具体负责政策执行的机构,涉及执行人员的情况以及领导者的领导模式、技巧等。再次,目标群体是指政策执行的实施对象,即受政策影响最大的群体。最后,政策环境是指与政策执行有关联的影响因子,包括政治、经济、社会、文化因素等。
1.2 药品上市后安全性研究政策执行分析框架
史密斯认为,政策执行过程是上述4 种变量之间互相作用、互相协调的过程。在此过程中,无论是政策参与者还是执行者,都会受到过程变量的影响,这两者之间的政策张力以及因此带来的冲突可能会引发“紧张”的政策关系。针对这种“紧张”的政策关系,可利用制度化或非制度化的措施处理出现的过程性问题并进行问题反馈,通过协调与修正变量之间的关系,使政策得以有效执行。基于史密斯模型中的4 个影响因子,构建PASS 政策执行分析框架,如图1 所示,这4 个影响因子会对PASS 政策执行产生一定的影响。
2、药品上市后安全性研究政策执行影响因素分析
2.1 理想化政策
2019 年新修订《药品管理法》实施后,MAH 制度开始在全国范围实施[7]。MAH 制度实施前,药品上市后阶段涉及药品流通质量管理、上市后监测、风险管理、批准后研究等,其责任主体包括药品生产企业、经营企业、医疗机构等,但未规定研发者对上市后药品的义务和责任。MAH 制度实施后,药品上市后阶段的责任主体为MAH。根据《药品管理法》第七十七条,MAH 应当制定药品上市后风险管理计划,主动开展药品上市后研究,对药品的安全性、有效性和质量可控性进行进一步确证,加强对已上市药品的持续管理[4]。2020 年新修订《药品注册管理办法》进一步细化了MAH 制度的框架、适用范围等。MAH 制度的实施,降低了药品研发机构的成本投入,激发了药品研发积极性。然而,MAH 主动收集不良反应并报告的意识仍有待提高,作为药品安全“第一责任人”,部分MAH 未贯彻落实《药品管理法》相关规定[8]。
GVP 是我国建立药物警戒制度以来出台的第一个药物警戒规范性文件, 论述了PASS 相关内容。但与美国、日本等国家相比, 我国对于PASS 还没有详细的规定。《药品管理法》第一百三十四条规定,MAH未按照规定开展药品不良反应(adverse drug reaction,ADR)监测或者报告疑似ADR的,责令限期改正,给予警告;逾期不改正的,责令停产停业整顿, 并处十万元以上一百万元以下的罚款。可见,目前我国对MAH 违法行为的处罚措施限于警告和罚款, 对MAH 的激励、警示和威慑作用仍有待加强。
2.2 执行机构
PASS 政策的执行机构主要是国家药监局、省级药品监管部门以及卫生行政部门等。药品监管部门和卫生行政部门的工作职责包括保障公众用药安全,并对ADR 监测和报告进行监督和管理。药物警戒贯穿药物发展全过程,包括上市前和上市后阶段,其中上市后监管包括ADR 监测和报告。MAH 制度实施前,我国药物警戒工作存在一些不足,如相关法律法规不完善、信息系统不成熟、药物警戒体系不健全等[8],ADR 报告和监测以被动为主,对于新的、严重的ADR 分析能力有限,药物警戒相关法律法规制定刻不容缓;MAH 制度的实施为建立健全药物警戒相关法律法规提供了依据,促进了我国GVP 的出台,推动我国药物警戒发展进入新阶段。同时,MAH 在ADR 监测和报告方面的主动性也有所提高,但其主动性意识仍需进一步加强。
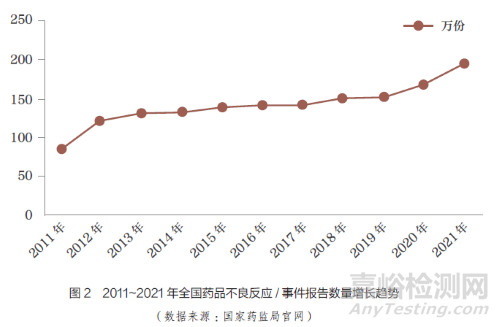
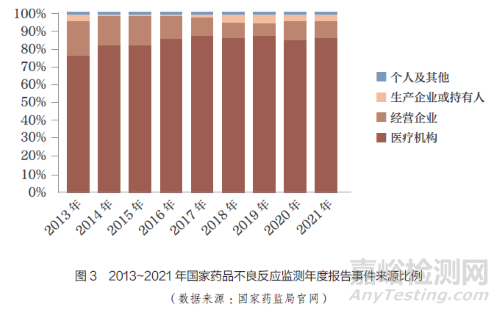
随着MAH、医疗机构以及患者对ADR 的重视,ADR 事件报告的数量逐年增加。药品不良事件是指在药物治疗过程中发生的任何不幸的医疗卫生事件,而这种事件不一定与药物治疗存在因果关系[9]。根据国家药监局统计,1999~2021 年, 全国ADR监测网络累计收到《药品不良反应/ 事件报告表》1883 万份[10],且每年ADR 事件数量不减反增,2011~2021 年全国不良反应/ 事件报告数量增长趋势如图2 所示。2021 年,全国不良反应/ 事件共196.2 万份,较2020 年增长17.1%,但来源于MAH 的报告数量仅占同期总数的4.1%,较2020 年的3.9% 并没有明显增长。由此可见,我国对ADR 的监测亟需进一步加强。2013~2021年,国家药监局发布的国家ADR监测年度报告来源情况如图3 所示。数据显示,近10 年来,医疗机构报告的ADR 事件占大多数,其次是药品生产经营企业;近3年来,MAH 报告的ADR 数量虽有所增加,但增幅不大,报告数量所占比例仍较低,我国GVP制度的落地还需要不断完善相关配套制度和规范。
在MAH 制度实施前,ADR报告的责任主体是药品生产和经营企业、医疗机构、个人及其他,但从ADR 报告的数量来看,药品生产和经营企业报告数量较少,主要原因在于企业追求利益最大化,不愿在ADR 监测和报告中投入更多人力、物力和财力,部分企业认为公众知晓ADR 报告会影响产品销量和企业形象。相信随着MAH 制度的持续推进,上述状况将得到改善。
2.3 目标群体
目标群体是指政策执行的实施对象。PASS 实施对象包括MAH、医疗机构及药品生产经营企业等。郭莎莎等[11] 认为,河北省仅有少部分MAH 开展风险评估,原因可能是MAH 把更多的资源和精力投入到药品上市前研究阶段,而减少了药品上市后风险管控的投入。方乐敏等[12] 认为,上海市部分MAH 存在未对药品安全数据进行回顾性评价、未实施评价和控制等问题,即使有些MAH 发现药品存在风险信号,也没有进行控制,无法实现风险管理的闭环。目前,我国ADR 报告来源以医疗机构为主,药品生产经营企业为辅,存在收集渠道不健全、漏报瞒报、信息利用率低、主动监测意识不足等问题[13]。而药物警戒制度的核心内容是药品风险管理,MAH 需要探测药品全生命周期中的隐患信号,在未发生严重ADR 事件时就加以控制,确保药品上市前与上市后的安全性。
药品生产企业是保障药品安全的责任主体。部分药品生产企业对药品上市前安全性研究的投入比上市后多,认为药品上市后安全性研究投入会增加企业负担。企业加大对药物警戒的资金投入,可能不会给企业带来直接的经济利益, 但可以保障企业获得更大的经济利益。药品生产企业有必要对上市后的药品进行监测, 降低药品上市后的安全性风险。
2.4 政策环境
我国药品监管由国家药监局、省级药监局、市县药监部门三级管理[4]。由于各省份、城乡之间医疗水平不同、资源分布不均、监管能力存在差异等,加之跨省监管协作机制仍有待完善,MAH制度顺利实施的难度较大[14]。目前,我国药品监管部门对药品的审批更加严格,并加强了上市后监管。然而,每年ADR 报告数量仍在增加,原因之一是ADR安全性评价机制仍有待完善。少数医疗机构为了通过审批伪造材料, 加之审批时间缩减, 导致药品审批监管难度加大, 药品质量和安全性受到影响[15-16]。新药上市前安全性研究需要经过临床试验阶段,试验会受到时间、用药人群等影响,导致新药上市后的风险不能被准确估量;而随着上市后用药人群的扩大,风险隐患也在增加。因此,相关部门有必要开展PASS 研究。
随着人们生活质量的提高,医疗卫生需求日益增加, 国家出台了一系列保证药品质量和安全的政策, 同时加深了人们对药品安全的认识。GVP 强调了在开展PASS 过程中的风险评估和控制,如MAH 应当监测PASS 期间的安全性信息, 发现任何可能影响药品获益- 风险平衡的新信息, 应当及时开展评估,并采取适宜风险控制措施;研究中发现可能严重危害患者生命安全或公众健康的药品安全问题时,MAH 应当立即采取暂停销售和使用、召回等紧急控制措施,并将召回和处理情况向省级药品监管部门和ADR 监测机构报告[6]。为了确保受试者或用药者的安全,MAH 要在整个PASS 过程中贯彻风险管理理念, 并对药品全生命周期进行监测。
3、药品上市后安全性研究政策执行建议
3.1 完善PASS 政策,出台PASS 管理规范和条例
我国的PASS 法律体系是以法律为主、法规为辅,共同指导MAH 进行PASS,同时由相关部门指导和监督研究全过程。MAH 应建立药品安全体系,对药品上市后安全进行监测,包括ADR 监测与报告流程规范、药品上市前后风险管理相关文件、药品上市后再评价相关规定等[16]。MAH 制度的不断完善,有利于MAH 开展PASS。
如上所述, 目前我国在PASS 政策执行过程中,对违法行为的行政处罚方式仅是警告和罚款。为保证PASS 政策的实施,建议在药品监管相关法律法规中对PASS 作出更详细的规定,对违法行为制定更严厉的处罚措施(如停止销售相关产品、吊销《药品生产许可证》等),以进一步督促MAH 自觉遵守PASS 相关规定。同时,建议相关部门出台PASS 相关管理规范等,从而更好指导MAH 开展PASS。
3.2 加强MAH 责任落实及对PASS 的监管,加大PASS 投入
MAH 是药物警戒的责任主体,职责包括对ADR 进行监测和报告。从已发生的ADR 事件来看,开展ADR 监测不仅有助于发现上市前受环境等因素影响未被发现的安全性问题,还有助于发现产品在流通或用药过程中出现的问题。同时,ADR 监测和报告的结果可作为药品安全性评价的依据,从而保障药品质量和公众用药安全。此外,药品生产经营企业在开展ADR 监测时,还可向公众展示最新的产品信息,这种具有主动性、自觉性的做法有利于企业树立良好形象,增加公众对企业的信任。因此,从长远来看,加大对PASS 的投入有利于企业发展。
3.3 提高药品上市后的风险管理
在PASS 政策执行过程中,需要进行药品上市后风险管理,包括识别药品风险信号、启动风险管理计划、药品安全性再评价、风险效益再评价等[17]。MAH 实施药物警戒制度时需改变思想观念,由被动转变为主动监测、识别、评估与控制药品风险。在监测阶段,MAH 应主动收集药物警戒相关信息并按要求上报,完善ADR 网络信息系统和药品监测机制,全面监测药品风险。在识别风险阶段,MAH 应增强对监测信息的分析能力,识别信息中是否存在潜在风险信号。在评估阶段,MAH 应重点关注具有潜在风险的信息,研究风险发生机制,定期对药品开展风险评估,包括药品安全性评价、药品潜在风险识别、主动开展PASS 等。在控制阶段,MAH 应根据风险评估结果,对存在的风险采取相应的解决措施,并撰写年度ADR 报告,减少药品对公众健康的危害。
3.4 健全药品上市后安全性评价体系
ADR 数据信息是开展药物警戒工作的基础。2016 年,原国家食品药品监督管理总局药品评价中心开发了中国医院药物警戒系统(CHPS),通过该系统可主动监测是否存在ADR 风险。该监测手段灵敏度高、便于数据分析,优化了医疗机构上报ADR 事件的流程,提高了医疗机构对ADR的监测水平,为药品上市后安全性评价提供了数据信息支撑。
药品上市后安全性评价体系的构建和完善是PASS 的内容之一,对于开展PASS 具有重要意义。药品上市前和上市后都需要进行风险评价,上市后安全性评价有利于识别和确认药品上市后是否存在安全隐患,如存在安全隐患,则需要采取暂停销售、召回药品等措施,从而有效防止更严重的ADR 事件发生。在药品审批阶段,建议药品监管部门加强对申请资料的审查,确保资料真实可靠。为保证安全性评价的真实性和准确性,建议选择德才兼备、经验丰富的医师作为药品安全性评价专家。此外,建议药品监管部门完善新药上市后安全性评价体系,定期对药品开展抽检和ADR 监测[18]。
近年来,我国不断出台规范药物警戒相关文件,药物警戒体系逐渐完善,相关法律法规不断成熟,为MAH 开展PASS 提供了保障。MAH 制度虽已被写入《药品管理法》中,但仍处于推进阶段,还需要不断完善。GVP 的颁布,促进了MAH 制度和PASS制度的推行,但同时需要注意,ADR 监测和管理方面还需完善,需要出台PASS 管理规范等来进一步明确PASS 相关内容,加强对PASS 的监管和风险管理,建立健全药品上市后安全性评价体系,从而最大程度地降低药品安全风险,保障公众生命安全与健康。