前 言
冷冻干燥是延长药品保质期的重要工艺手段;
到2015年,冻干注射剂产品占所有新的注射剂药品的近一半;
本篇文章将FDA从2015年 - 2019年收到的263个冻干注射剂产品(创新药和仿制药)中确定的制造相关缺陷进行分类;
从配液、过滤、灌装、冻干四个工艺阶段,对缺陷进行分别的讨论;
从监管视角,为大家提供提供一份常见缺陷清单;
希望本系列文章提供的信息,可以帮助大家避免这些与制造相关的常见缺陷;
从而加速高质量冻干产品的审批和上市流程;
本文在此仅讨论OPMA收到的监管报告中确定的与制造商相关常见缺陷;
仅为申请人每个单元操作中常见缺陷;
并非对申请人需要进行的潜在研究进行全面讨论;
为避免FDA发布大量缺陷,申请人应明确提供产品开发过程的详细信息,以及如何利用开发数据和风险评估来减轻与商业制造过程相关的任何风险;
数据来源和研究方法:
从2015年到2019年,共收到263份冻干注射产品的规范提交文件,并在FDA的内部数据库中搜索与制造相关的缺陷;本研究调查了原始申请和修正案;根据生产冻干注射产品所需的常用操作流程,OPMA发布的缺陷分为四类:混合、过滤、填充和冷冻干燥;每一类别缺陷以占与制造相关缺陷总数的百分比表示;文中还提供了FDA发布的每个类别的缺陷示例(文中灰色底色部分);需要注意的是,在过滤和下游过程中观察到的与无菌保证相关的缺陷超出了本工作的范围。
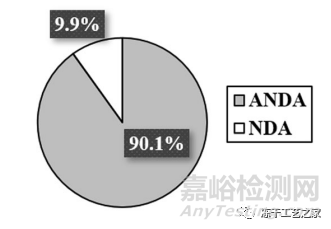
FDA从2015年 - 2019年收到的263个冻干注射剂产品(创新药和仿制药)
图一:冻干注射剂提交文件中的制造相关缺陷
(在263份提交文件中约占83%,包括199个ANDA和19个NDA)
图二:在多个操作单元观察到多个缺陷
(缺陷占比之和,超过100%)
配 液
配液开始时,完成初始物料的分配;
对于高风险、容易降解的物料,应在批次记录明确环境控制参数(例如温度、相对湿度、惰性气氛和光照),以减少操作过程中的降解。
例如:为了避免原料降解,在低温条件下存储的材料,需要控制操作环境条件和操作时间之间的平衡。
应采用最佳的称重策略,以尽量减少称重的不准确。
根据21CFR 211.101(a)规定:API总量不应低于标记量的100%。
为了达到这个目标,通常会根据效力的变化(例如,HPLC测定、水含量和残留溶剂)调整API的重量。这对于纯度较低的API(如由于盐的存在或者残余水分影响)或会因为在存储期间不稳以及由于吸湿性会导致效力降低的API来说,尤为重要。
因为以上原因在配液过程中导致的过量投料,必须解释过量投料的原因以及过量投料具体的量。
根据ICH Q1A(R2),申报批次生产过程中应该用不同批次的API分别验证。将API批次混合在一起进行生产的做法妨碍了对单一批次原料对制剂影响的考察。这种做法不符合ICH指南的意图。
配液过程中,API和赋形剂混合均匀。
药品配方的PH经常进行调整,以使得API的溶解度和稳定性达到最佳值;
该步骤的典型开发过程包括但不限于:
1、 原辅料的添加顺序;
2、 API在不同温度和PH范围内的溶解度和稳定性;
3、 半成品制剂暴露于氧气或光照条件下的稳定性;
4、 正常生产条件下API的溶解速率;
这些研究为指定生产工艺控制条件提供依据,如:温度、PH、溶氧水平、最长处理和保持时间、最终制剂的API目标浓度。
配液环节常见缺陷是缺少对以上参数的控制以及缺少足够的放大数据支持性研究。
这些研究对于溶解度优先、需要较长溶解时间的API尤其重要。
过程控制应至少包含对API和辅料完全溶解的目视检查。
根据药物制剂的成分和确保药品质量的总体控制策略,可能还需要额外的(化验、PH值、有关物质)检验。
对于胶体分散体、悬浮液和乳液等非单相体系制剂,需要关注配液过程中混合的均匀性。
对于处方中含有金属螯合剂或者强酸的产品,所有和药液接触的金属材质需提供符合ASTM 316/316L要求的证明文件,以降低元素杂志和表面腐蚀的风险。
与配液有关的常见缺陷示例
如果过量投料超过3%,如果这个过量投料是为了补偿制造过程中的损失。提供具体的控制策略,以确保不同批次的损失是一致的,并且最终的药品将会符合规范;
溶解性差的原料,申报批的搅拌过程为15小时。商业批批量是申报批的8倍,但没有提供确保API完全溶解的控制策略。需证明商业规模工艺参数(搅拌速率、搅拌时间等)足以确保API充分溶解。另外,我们注意到药品溶解后外观浑浊,阻碍了通过目视检查来验证溶解的方法。建议在过滤后对药品进行化验检测,作为过程控制的数据;
药品处方中含有EDTA,这是一种金属螯合剂,提供文件证明与药液接触的金属设备符合ASTM对316/316L的要求;
半成品制剂PH范围太宽并未提供制定依据,请缩紧PH接受限度或证明原料在这个PH范围内的溶解性和稳定性都能满足要求;
申报批中,配液过程每个添加步骤后的搅拌时间没有明确规定终点,而是规定操作员“搅拌至溶解”或者“搅拌时间不少于一定时间”。建议根据开发过程数据或者申报批的生产数据来确定搅拌时间的上限和下限;
无菌过滤
通常采用过滤来确保冻干前药品制剂的无菌性。
众所周知,某些API和赋形剂(例如,防腐剂、有机溶剂和清洁剂)可能被过滤膜以及过滤过程中使用的管路吸收,从而降低效力,也会降低防腐剂的有效性。
此外,当过滤器被配方以外的液体润湿时(即,在预过滤完整性研究期间),通过过滤器的初始散装产品可能会被过滤器中残留的润湿剂稀释。
鉴于这些情况,如果未建立最小冲洗量且未提供证明冲洗这些风险的化验/防腐剂浓度数据可能会导致缺陷。
值得一提的是,进行相容性测试以证明本体溶液的化学稳定性(例如含量、相关物质和pH值)。
此外,需要根据21 CFR 211.65和ICH Q7 5.1评估与药品配方接触的所有制造组件(例如,储存容器、管路和过滤器、垫圈)的化学安全性。
最常见的缺陷之一是评估不足,无法降低与药品配方接触的制造组件的潜在可提取物和可浸出物相关的风险,尤其是含有有机溶剂、表面活性剂或螯合剂的配方。
可以参考USP以及可提取物和可浸出物研究的一般原则和最佳实践建议,其中包括用于设计和证明适当研究的有用信息。
产品符合USP要求,可能足以降低水性配方中可浸出杂质的风险。
同样重要的是,对于较高风险的配方和/或制造条件,需要对可提取物/可浸出物进行完整的化学安全评估。
虽然上述一些 USP 章节主要针对容器/密闭系统,但其中的原则和方法同样适用于与药品制剂直接接触的所有制造设备。
与无菌过滤相关的常见缺陷示例
需要在过滤药品制剂之前,定义最小冲洗体积,以降低API 吸附或药品制剂稀释相关的风险。
提供文件证明在制造过程中与药品配方接触的聚合物成分(过滤器、管路等)符合CFR非直接食品添加剂的要求以及USP要求。
药品配方中含有大量用于溶解API的环糊精,这也增加了聚合物制造设备(过滤器、管路等)中可浸出物杂质的风险。提供特定设备最差制造条件下的可提取研究。或者,也可提供最终药品的浸出物研究。
用水作为提取溶剂,对除菌过滤器进行可提取物研究;然而,由于药品配方中有高含量的有机溶剂,因此这种溶剂选择并不合适。使用与药品配方具有相似提取能力的模型溶剂,提供修改后的可提取研究。
目前,我们的毒理学团队正在审查你们在可提取物研究中确定的来自制造设备的潜在浸出物的毒理学说明。我们鼓励您提交对最终药品的可浸出物研究,以确认或否定可浸出物或其衍生物的存在水平高于分析评估阈值
灌 装
过滤完成后根据配方灌装。
申请人需要确保过滤后的药品制剂在灌装前保持完整的化学稳定性。
产品的最大保持时间应根据产品特性(如化学、物理或微生物数据,或其他适用数据)拟定。
此外,申请人应说明对灌装过程中暂停的时间上限(如有)的理解,特别是在可预见对原料药的稳定性和效力造成风险和/或可浸出物增加的情况下。
需注意,USP<1151>中规定的过量灌装体积不建议用于冻干产品的装量设定。相反,USP<1151>中的过量灌装体积建议适用于重组药物产品的灌装,数据可以通过实验确定。
关于灌装量的上限,灌装过程应适当的提供可接受的过量灌装,以满足标签上标注的要求的正确的剂量。
灌装量控制策略应确保每一支西林瓶装量都在检测规范的可接受标准范围内,并确保符合USP<905>。
因此建议申请人提供下列信息,使得可以根据标签上的信息进行正确的给药:
(1)药品制剂的浓度;
(2) 指定的装量范围;
(3) 根据标签,用于重构的稀释剂的体积;
(4) 复原产品的总体积;和(5)根据标签,最终重组产品的给药量,说明在提取和给药过程中的损失。
在灌装过程中,申请人应通过建立100%在线称重方式或详细的抽样计划(例如样本量和频率)来证明对灌装量的充分控制。
与灌装相关的常见缺陷示例
提供按照标签要求的重组药品的总量。如果总量少于USP<1151>的建议,请按照标签进行停药和给药研究,并遵守USP<697>中制定的指南。报告测得的可注射量。
基于重组药物产品的总量,需要进行超量灌装以达到0.5mg API/mL重组药物需求浓度。调整您的灌装量控制策略,以确保产品达到标示浓度,从而确保患者的正确剂量。
提供灌装过程中的在线灌装重量检查的取样计划详情。
暂停灌装会使输送管线中的药物制剂升温至室温。您已经表明,药物制剂中的原料药在室温下保存时对降解很敏感。提供灌装过程中的停止时限,以及重新开始灌装操作时的最小排空量,以确保药品质量。验证您的控制策略。
药物制剂对光诱导和氧化降解敏感。澄清已灌装西林瓶在装载到冻干单元之前的储存区域控制策略,以确保药品质量
生产时间
冻干注射剂在NDA和ANDA审查期间的一个常见缺陷与生产操作时长限度的制定有关(表V)。
根据21 CFR 211.111条的规定,生产过程的每个环节都应该有时间限制。
从API投料开始的每个环节的暂停和保持时间,都应该有数据支持(如,化学、物理或微生物数据,以适用者为准)。
与生产时间相关的常见缺陷示例
API在溶解的过程中容易水解、降解,高温会加速水解和降解。因此需要确定从配液过程中添加原料到冻干开始的最长时间限制。证明中间体在和生产设备接触后,在最坏的制造参数下(时间、温度等),产品质量仍可符合目标要求。
处方中含有有机溶剂,在生产过程中与聚合物成分接触(过滤器、管道等)会增加可浸出杂质的风险。在灌装过程中应制定最大暂停时间,以限制中间体和生产设备的静态接触。并在重新开始灌装时提供最小冲洗量。并证明这些控制参数能降低最终药液和生产设备接触导致终产品浸出杂质的风险;
收 率
在所提供的商业生产批次记录中,缺乏对产品收率的控制不能满足 21 CFR 211.103,211.118(b)和211.186(b)而导致的缺陷,。
制定产品收率限制保证了当产量出现无法解释的差异时,能够去调查这个差异对药品质量和安全性问题的影响。收率限制的制定确保符合21CFR 211.192要求。
通常会发现,批次记录中没有定义实际的产量限制,缺乏控制上限,或者范围过宽,无法反应关键生产过程。
在工艺验证批时应确定验收收率范围。
然而,在申报资料提交时,都是根据可用的注册批和稳定性批数据制定暂定的收率范围限制。
与收益率相关的常见缺陷示例
在审查主批次记录(MBR)时,发现批次记录中没有规定收率范围。请根据之前生产批次情况,为每个生产步骤制定收率限制范围以确保工艺的稳健性,并在整个制造过程中充分考虑使用的材料情况,根据这个更新MBR。
冷冻干燥生产步骤实际产量“不低于40%”的验收标准不受以下结果的支持:注册批次平均为(98.0±2.0)%。此外验收标准未遵循21 CFR 211.186(b)制定严格的收率标准范围(上下限);
批 量
制剂稳定性批次的批量大小应足够建立工艺参数。
尽管ICH指南没有明确定义,但在2014年FDA行业指南相关问题的回答文件中讨论了最小稳定性批次大小。
在药物申请文件中偶尔会观察到稳定性批次执行过程中未能达到最小批量预期。
上述FDA指南中提供了与目标西林瓶灌装量相匹配的批量建议。偏离这些指导建议需要适当的理由。
与批量相关的常见缺陷示例
根据您确定的灌装体积8ml,建议注册稳定性批次的最小批量为30L.我们注意到您的注册批量为7.5L,您计划扩大到75L的商业批量。我们建议您修改您的商业批量大小与注册批量相同,并提供一个确认:在未来扩大时将提供事先批准补充(PAS)。或者,您可以提交来自适当批量大小的新的注册批次数据。请参考FDA行业指南“仿制药:原料药和成品的稳定性测试—问答”
目 检
一般来说,蛋糕外观是冻干药品的关键质量属性之一。
有了明确的粉饼外观验收标准,应采取适当的工艺控制措施,以确保批内和批间粉饼外观无显著差异。
目检是冻干操作中的一个关键控制策略,因为蛋糕外观的异常可以提醒您制造过程中的问题。这些问题可能与产品质量属性有关,也可能与产品质量属性无关。
在检查生产设备的过程中,通常会对目视检查实践进行审查。
建立目视检查程序的指南可以在USP <790>和<1790>中找到。
在对申请进行审查期间,出现的两种常见缺陷如下:
(1)未能建立典型的目视检查拒收率或数量限制,超过限制将会引发对批次的调查;
(2)未能对注册批次中观察到的较高比例目视检查拒收进行解释和纠正措施(表八)。
在原始提交文件中解决这些问题将有助于证明有足够的控制措施来检测和解决冻干药品中的潜在质量缺陷。
与目检相关的缺陷常见缺陷示例
更新商业生产批报告(MBR)中母舰的不合格品率;
注册批目检的不合格率高达12%,解释原因并制定商业批如何减少不合格率的控制措施。